{kind=link}
{kind=link}
{kind=link}
Salen-Mn(Ⅲ)和Salen-Mo(Ⅳ)配合物在介孔硅胶上接枝及烯烃环氧化催化性能
引用本文
周建波, 徐超, 曾明, 阳科, 崔小莹. Salen-Mn(Ⅲ)和Salen-Mo(Ⅳ)配合物在介孔硅胶上接枝及烯烃环氧化催化性能[J]. 工业催化, 2015,23(10): 767-772.
Zhou Jianbo, Xu Chao, Zeng Ming, Yang Ke, Cui Xiaoying. Salen-Mn(Ⅲ) and Salen-Mo(Ⅳ) complexes immobilized on mesoporous[J]. Industrial Catalysis, 2015,23(10): 767-772.
DOI:10.3969/j.issn.1008-1143.2015.10.007
Zhou Jianbo, Xu Chao, Zeng Ming, Yang Ke, Cui Xiaoying. Salen-Mn(Ⅲ) and Salen-Mo(Ⅳ) complexes immobilized on mesoporous[J]. Industrial Catalysis, 2015,23(10): 767-772.
Permissions
Copyright©2015, 《工业催化》编辑部
《工业催化》编辑部 所有
Salen-Mn(Ⅲ)和Salen-Mo(Ⅳ)配合物在介孔硅胶上接枝及烯烃环氧化催化性能
作者简介:周建波,1979年生,女,湖南省衡阳市人,讲师,主要从事不对称催化合成研究。
摘要
通过肽键作用将Salen型金属配合物接枝到介孔硅胶孔道中生成固相催化剂,采用红外光谱、热重分析和元素分析等对制备的固相催化剂进行表征,结果证实,Salen型金属配合物成功接枝到介孔硅胶载体上。以环辛烯和环己烯为反应底物,叔丁基过氧化氢和过氧化氢为氧化剂,比较均相Salen型催化剂和多相Salen型催化剂的催化活性。制备的均相Salen型催化剂和利用肽键键合制备的固相催化剂均具有一定的催化性能,Mo-Salen催化活性更高,是因为叔丁基过氧化氢在Mo-Salen存在下易分解。固相催化剂活性≤75 ℃时稳定,在氧化剂叔丁基过氧化氢和过氧化氢作用下具有较好的稳定性能。重复实验中,金属离子流失量很小,催化活性和TOF值未降低,表明利用肽键制备的固相催化剂催化活性稳定,为固相催化剂的制备开辟新思路。
关键词:
催化剂工程; Salen-Mn(Ⅲ); Salen-Mo(Ⅳ); 介孔硅胶; 肽键固载; 环氧化
中图分类号:TQ426.6
文献标志码:A
文章编号:1008-1143(2015)10-0767-06
Salen-Mn(Ⅲ) and Salen-Mo(Ⅳ) complexes immobilized on mesoporous
Abstract
The metal Salen complexes were immobilized on the organo-modified silica framework by peptide bond interactions.The immobilized Salen complexes were characterized by FT-IR,TG-DTA and elemental analysis.The results confirmed that salen-Mn(Ⅲ) and Salen-Mo(Ⅳ) complexes were attached to the silica framework.Using tert-butyl hydroperoxide and hydrogen peroxide as the oxidants,the catalytic performance of homogeneous catalysts and immobilized catalysts for catalytic epoxidation of cyclooctene and cyclohexene were studied.The homogeneous and immobilized Mo-Salen complexes exhibited higher catalytic activity for the epoxidation,whereas Mn-Salen complex systems caused additionally non-productive decomposition of tert-butyl hydroperoxide.The immobilized catalysts had good and stable activity below 75 ℃ and in the existence of tert-butyl hydroperoxide and hydrogen peroxide.In repeated experiments,the immobilized Salen complex was resistant to leaching of Salen molecule,and its catalytic activity and TOF did not reduce,which indicated that the immobilized catalysts had stabe catalytic activity.
Keyword:
catalyst engineering; Salen-Mn(Ⅲ); Salen-Mo(Ⅳ); mesoporous silica gel; peptide immobilization; epoxidation
为合成高效和实用的生物活性物质出现许多不对称化学反应, 其中, 烯烃环氧化产物在有机合成和药物合成中得到广泛应用[1]。为了使烯烃环氧化反应高效、安全和高选择地进行, 对过渡金属配合物作为环氧化反应催化剂[2]进行研究, 合成经典手性席夫碱Salen-Mn均相催化剂, 并开发Salen-Fe和Salen-Mo等催化剂, 这类催化剂具有高活性和高立体选择性等优点, 但存在难分离回收、难重复使用和在反应中容易二聚失活等缺点。为避免以上缺点, 试图将Salen配合物利用化学接枝或包络合法固载到不溶高聚物[3, 4]、中孔分子筛[5]、MCM-41[6, 7]、黏土[8]和硅胶[9]等固相载体上制备出固相催化剂。化学接枝制备的催化剂稳定性高, 重复使用性能好。
通常通过共价键将Salen-Mn(Ⅲ )和Salen-Mo(Ⅳ )配合物接枝到分子筛上生成稳定的固相催化剂。本文利用范德华力中的肽键将Salen-Mn(Ⅲ )和Salen-Mo(Ⅳ )固载到有机物修饰的介孔硅胶载体上成功制备出环氧化固相催化剂, 采用红外光谱、热重分析和元素分析等进行表征, 以环己烯和环辛烯为反应底物、叔丁基过氧化氢和过氧化氢为氧化剂, 考察制备的固相催化剂催化活性, 并与均相催化剂进行比较, 回收实验验证所得固相催化剂的重复使用性能。
1 实验部分
1.1 仪器与试剂
自动控温电磁搅拌器, 常规玻璃仪器, 索式提取器, 真空干燥装置, 马弗炉, Nicolet 510P型FT-IR 红外光谱仪, 德国耐驰公司NETZSCH STA 409 PC TG-DSC仪, 美国安捷伦公司Agilent 1100型高效液相色谱仪, 德国Vario ELⅢ 元素分析仪, 美国热电公司ICAP6500DOU型电感耦合等离子发射光谱仪。
3, 5-二叔丁基水杨醛, 3, 4-二胺苯甲酸, 二氯化锰, 无定形介孔硅胶, 过氧化氢, 叔丁基过氧化氢, 环己烯, 环辛烯, 二氯化锌, 1-羟基-苯并-三氮唑, (3-氨基丙基)三甲氧基硅烷, N, N'-二环己基碳酰亚胺, 六羰基钼, 有机溶剂, 试剂均为分析纯。
1.2 均相Salen-Mn(Ⅲ )配合物合成
依据文献[10]将20 mmol的3, 5-二叔丁基水杨醛和10 mmol的ZnCl2溶解在40 mL的四氢呋喃中, 将其逐滴滴加到含有10 mmol的3, 4-二胺苯甲酸的50 mL四氢呋喃中, 冷凝回流45 min, 冰水冷却, 抽滤, 25 mL甲醇洗涤, 冷却结晶得到黄绿色Salen配体。
将Salen配体溶解于70 mL含有1.2 mmol的MnCl2四氢呋喃溶液中, 得黄棕色溶液, 室温搅拌1 h, 冷凝回流0.5 h, 冷却至室温, 缓慢加入2.4 mmol三乙胺, 通入45 min空气, 减压蒸馏去除溶剂, 剩余物用20 mL甲苯洗涤, 搅拌, 过滤, 减压蒸馏除去甲苯, 即得黑色泡沫固体Salen-Mn(Ⅲ )配合物, 标记为A1。
1.3 均相Salen-Mo(Ⅳ )配合物合成
将2 mmol的Mo(CO)6溶解于100 mL四氢呋喃, 加入2 mmol制备的Salen-Mn(Ⅲ )配合物得浅棕色溶液, 回流24 h, 过滤, 减压蒸馏除去溶剂, 得到黑色泡沫固体产物, 标记为A2。
1.4 有机功能化硅胶制备
有机功能化硅胶制备参见文献[11]。5 g不定型硅胶粉末70 ℃真空干燥箱中干燥3 h, 将其分散到90 mL甲苯中, 加入28 mmol的(3-氨基丙基)三甲氧基硅烷, 磁力搅拌下加热回流10 h, 所得悬浮物真空过滤得沉淀物, 沉淀物在索氏提取器中用异丙醇洗涤24 h, 100 ℃干燥5 h, 得到有机物改性硅胶B。
1.5 Salen配合物固载化
将A1或A2溶解于30 mL二氯甲烷, 1 mmol的1-羟基-苯并-三氮唑分批加入其中, 强力搅拌10 min, 加入3 mL溶解5.0 mmol的N, N'-二环己基碳酰亚胺的CH2Cl2溶液, 室温搅拌40 min, 1.3 g有机功能化硅胶(约1.87 mmol, 75 ℃真空干燥)缓慢加入到混合物中, 混合物在磁力搅拌下冷凝回流反应15 h, 过滤得固体物, 固体物在索氏提取器中分别用乙醇洗涤30 h, 丙酮洗涤10 h, 得到固相催化剂C1和C2。
1.6 催化剂表征
红外光谱检测在美国Nicolet公司Nicolet Nexus FT-IR红外光谱仪上分析, 样品与KBr混合压片, 摄取(400~4 000) cm-1红外光谱, 分辨率4 cm-1。
热分析在德国耐驰公司NETZSCH STA409PC TG-DSC仪上进行, 测定温度(273~1 073) K, 升温速率为10 K· min-1。
元素分析在德国Vario ELⅢ 元素分析仪上进行。
ICP-OES在美国热电公司ICAP6500DOU型电感耦合等离子发射光谱仪上进行, 功率1 150 W, 泵速60 r· min-1, 雾化器流量0.55 L· min-1, 冷却气流量14 L· min-1, 辅助气流量0.5 L· min-1。
一定量催化剂, 反应底物、氧化剂和溶剂在不同温度下磁力搅拌反应, 定时取样, 在Agilent 1100型高效液相色谱仪上分析产率和选择性。
2 结果与讨论
2.1 元素分析和ICP-OES
表1为氨基功能化硅胶B和催化剂C1和C2的元素分析及ICP-OES分析。
| 表 1 氨基功能化硅胶B和催化剂C1和C2的元素分析及ICP-OES分析 Table 1 Elemental and ICP-OES analysis of amino-functionalization silica gel B, catalysts C1 and C2 |
从表1可以看出, 有机功能化硅胶接枝的氨基丙基通过元素分析得到证实, 氨基丙基接枝量为1.44 mmol· g-1, 用ICP-OES检测C1和C2催化剂的Mn和Mo含量, Salen-Mn和Salen-Mo含量分别为0.22 mmol· g-1和0.16 mmol· g-1, 折算成Mn和Mo质量分数分别为1.2%和1.5%。通过氨基丙基改性的硅胶只有约14%的氨基接枝了Salen-Mn配合物; 12.5%的氨基接枝了Salen-Mo配合物。通过实验证实了分子筛表面功能化氨基不会引起环氧化物的开环反应。
2.2 TG-DTA
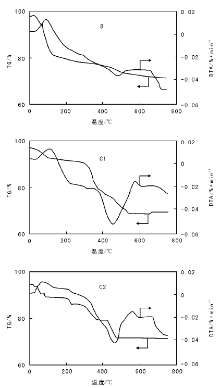
图1为氨基功能化硅胶B和催化剂C1、C2的TG-DTG曲线。
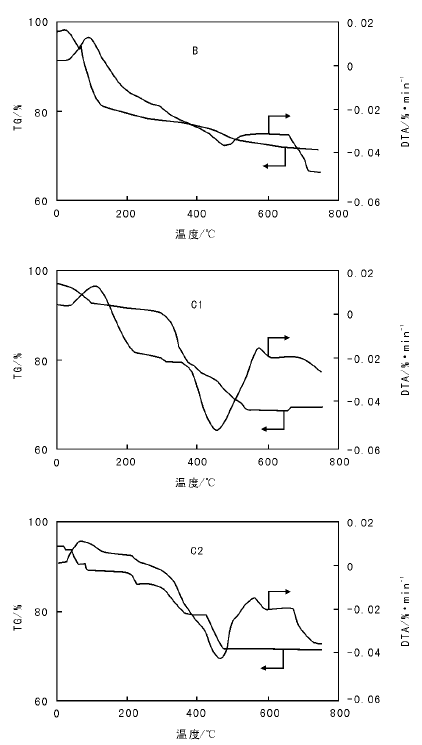 | 图 1 氨基功能化硅胶B和催化剂C1、C2的TG-DTA曲线Figure 1 TG-DTA curves of amino-functionalization silica gel B, and catalysts C1 and C2 |
从图1可以看出, C1、C2的DTA-TG相似, 而B差别较大。100 ℃时, B、C1和C2均有个明显的失重台阶, 对应一个吸热峰, 应该归结为样品中水的蒸发过程; (300~500) ℃, B、C1和C2的失重率分别约为8%、22%和19%, 与元素分析数据吻合, C1和C2的失重率大于B, 应该是C1和C2孔道中接枝的Salen配体失重的结果, 而且失重主要集中在(400~500) ℃, 温度较高, 说明Salen配体通过肽键接枝在硅胶孔道中, 而不是简单的物理吸附。
2.3 FT-IR
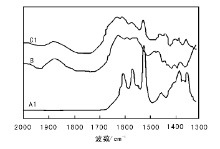
图2为氨基功能化硅胶B和A1、C1的FT-IR谱图。
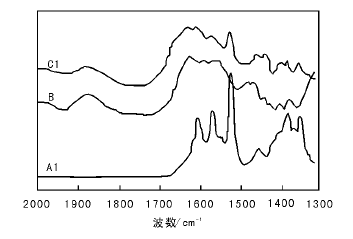 | 图 2 氨基功能化硅胶B和A1、C1的FT-IR谱图Figure 2 FT- IR spectra of amino-functionalization silica gel B, and A1 and C1 |
由图2可见, A1在1 534 cm-1、1 581 cm-1和1 614 cm-1处分别出现、和羰基的
特征峰, 在1 365 cm-1处出现C— O振动峰, B没有, 而C1在1 585 cm-1和1 620 cm-1处同样出现和特征峰, 在1 366 cm-1处出现C— O振动峰, 表明Salen-Mn配合物通过肽键成功接枝到氨基改性硅胶孔道中。另外, B和C1在1 100 cm-1、802 cm-1和470 cm-1处均显示出Si— O— Si特征峰, 1 101 cm-1和1 200 cm-1处出现Si— C振动峰。
2.4 环氧化反应
为了比较肽键接枝的固相催化剂和均相催化剂的催化活性, 在反应温度75 ℃、叔丁基过氧化氢为氧化剂和环辛烯为反应底物条件下, 分别以均相Salen-Mn和Salen-Mo、固相Salen-Mn和Salen-Mo为催化剂, 考察其在环氧化反应的催化活性, 结果如表2所示。
| 表 2 催化剂催化环辛烯环氧化反应结果比较 Table 2 Catalytic activity comparison of the homogeneous Salen-Mn and Salen-Mo catalysts and peptide immobilized Salen-Mn and Salen-Mo catalysts |
从表2可以看出, 均相催化剂和固相催化剂的催化活性差别不大, 反应0.5 h时, 固相催化剂的产率相对均相催化剂有小幅下降, 但随着反应时间的延长, 固相催化剂和均相催化剂活性渐趋一致。 可能是因为反应开始时, 反应底物要进入到催化剂孔道才能接近催化活性中心, 影响反应速率。Salen-Mo型催化剂比Salen-Mn型催化剂具有更好的催化活性, 因为Salen-Mn型催化剂容易使氧化剂分解。
2.4.1 反应温度
以Salen-Mn为催化剂, n(环辛烯)∶ n(叔丁基过氧化氢)∶ n(催化剂)=1∶ 3∶ 0.01, 考察反应温度对叔丁基过氧化氢分解率和环辛烯环氧化产率的影响, 结果见表3。从表3可以看出, 相同反应时间内, 随着反应温度升高, 叔丁基过氧化氢分解率提高, 产率先增后降, 原因是叔丁基过氧化氢在Salen-Mn配合物的影响下易分解, 这也是以Salen-Mn为催化剂时环氧化产率低于50%的原因。
| 表 3 反应温度对叔丁基过氧化氢分解率和环辛烯环氧化产率的影响 Table 3 Effects of reaction temperatures on decomposition rate of tert-butyl hydroperoxide and the yield of yclooctene epoxidation |
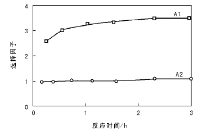
为了比较Salen-Mn和Salen-Mo在环氧化反应中的选择性, 考察不同时间选择因子。
选择因子=
图3为A1和A2在60 ℃的选择因子。从图3可以看出, A2的选择因子接近1, 表明氧化剂叔丁基过氧化氢100%参与了环氧化反应; 而A1的选择因子均为3, 表明Mn-Salen除了能催化环氧化反应还能引起叔丁基过氧化氢的分解, 只有三分之一的叔丁基过氧化氢作为氧化剂发生环氧化反应, 三分之二由于Mn金属配合物的存在而发生分解反应。
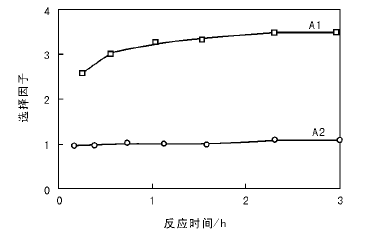 | 图 3 A1和A2在60 ℃的选择因子Figure 3 Selectivity factor SF at 60 ℃ during cyclooctene epoxidation in presence of homogeneous manganese A1 and molybdenum A2 complexes |
2.4.2 反应底物与氧化剂对C2催化性能的影响
考察不同反应底物与氧化剂对C2催化性能的影响, 结果见表4。
| 表 4 不同反应底物与氧化剂对C2催化性能的影响 Table 4 Catalytic activity of peptide immobilized Mo-Salen catalyst C2 for cyclohexene and cyclooctene epoxidation with hydrogen peroxide and tert-butyl hydroperoxide, respectively |
从表4可以看出, 相同反应条件下, 环辛烯比环己烯的环氧化产率略高, 与文献[12]一致。反应底物均为环辛烯, 叔丁基过氧化氢作氧化剂的环氧化产率比30%过氧化氢高很多, 表明30%过氧化氢中的过氧化氢对反应起抑制作用[13]。可能原因是有机过氧化物中的烷基基团比过氧化氢对反应底物和催化活性中心Mo具有更好的亲和作用; 另外H2O对分子筛表面具有强烈的覆盖作用, 阻塞了硅胶的表面孔道, 抑制环氧化反应的进行。
2.4.3 催化剂重复使用性能
在叔丁基过氧化氢用量5.1 mmol、环辛烯用量1.7 mmol、催化剂(金属活性中心)C2用量0.015 mmol、甲苯用量15 mL、反应温度75 ℃和反应时间1 h条件下, 考察催化剂C2的重复使用性能, 结果见表5。
| 表 5 催化剂C2的重复使用性能 Table 5 Repeated use times of peptide immobilized Mo-Salen catalyst C2 for cyclooctene epoxidation |
实验过程中发现, 第一次催化反应时, 催化剂颜色由深棕色转化成黄色, 可能是在环氧化过程中催化活性中心由Mo(+Ⅳ )转化成Mo(+Ⅵ ), Mo(+Ⅵ )比Mo(+Ⅳ )具有更好的催化活性, 催化剂在重复实验中的产率和TOF值均比第一次使用时略高。从表5可以看出, 催化剂第一次使用有少量的Mo-Salen配合物流失, 重复使用时流失量很小, 表明利用肽键成功将Salen配合物接枝到硅胶上, 所得接枝型催化剂性质稳定。
3 结论
(1) 催化环氧化结果显示, 制备的均相Salen型催化剂和利用肽键键合制备的固相催化剂均具有一定的催化性能, Mo-Salen催化活性更高, 是因为叔丁基过氧化氢在Mo-Salen存在下易分解。
(2) 考察不同反应温度和氧化剂对催化剂催化性能的影响, 固相催化剂活性≤ 75 ℃时稳定, 在氧化剂叔丁基过氧化氢和过氧化氢作用下也具有很好的稳定性能。重复实验中, 金属离子流失量很小, 催化活性和TOF值未降低, 表明利用肽键制备的固相催化剂催化活性稳定, 为固相催化剂的制备开辟了新思路。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|