







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
第二章:固体催化剂制备技术原理
引用本文
展恩胜, 李勇, 申文杰. 第二章:固体催化剂制备技术原理[J]. 工业催化, 2015,23(11): 932-960.

Permissions
Copyright©2015, 《工业催化》编辑部
《工业催化》编辑部 所有
第二章:固体催化剂制备技术原理
2.1 前 言
催化剂和催化反应过程广泛应用于能源转化利用、化学品制造和环境治理等领域。据估算, 约有85%的化学工业过程涉及催化反应过程, 因此催化也被认为是现代化学工业的基石。近年来, 提高催化反应过程效率和开发新催化反应已成为实现能源(资源)高效清洁利用和化学/化工生产绿色化的重要途径。催化过程的核心和物质基础是催化剂, 目前约有80%的工业催化过程采用固体催化剂(其余17%为匀相催化, 3%为生物催化)[1]。
固体催化剂的制备包含沉淀、浸渍、老化、过滤(包括洗涤)、干燥和焙烧等诸多单元操作步骤, 每一步均涉及复杂的物理、化学变化。催化剂制备技术在很大程度上影响催化剂的物理和化学结构, 并最终决定催化剂的性能(活性、选择性和稳定性)。长期以来, 催化剂的开发大多基于经验性积累, 因此催化剂制备也被认为是“ 技艺” 而非科学, 这些经验多是基于“ 试错法” 获得的规律性认识, 或是对催化剂制备过程特定操作单元和对催化剂结构特定参数的理解得到。例如在合成氨铁基催化剂开发过程中, 研究者筛选了不同组分和不同制备方式, 获得了约4 000多个催化剂样品, 进行了超过10 000次的实验才最终成功得到了获得工业应用的熔铁催化剂[2]。不可否认, 在催化科学特别是工业催化剂开发的历史上, 这种方式曾经而且仍然扮演着重要作用, 但由于缺乏可靠的科学基础作为依据, 这种方式往往经验性强、效率较低。自20世纪后期, 发展催化剂制备科学逐渐成为催化研究的重要方向, 也逐步与物理化学和材料科学融合交叉。
固体催化剂大致可以分为本体催化剂(bulk catalyst)和负载催化剂。本体催化剂是利用固体材料本身作为催化的活性相, 其特征是催化剂颗粒外表面和内部构成基本一致, 均具有催化活性, 常见的如雷尼镍等骨架合金型催化剂和分子筛催化剂; 负载催化剂是利用具有较大比表面积的多孔结构固体材料作为载体, 对活性相起分散和稳定作用, 这类催化剂的活性相多是贵金属或难以获得大比表面积的组分, 常见的负载催化剂如:Pt/分子筛加氢异构催化剂、Pt/Al2O3重整催化剂以及Pd/C加氢催化剂等。
近年来, 很多文献和论著对固体催化剂制备技术和原理进行了详尽总结[1, 2, 3, 4, 5]。本体催化剂和催化剂载体制备方法主要有沉淀/共沉淀法、溶胶-凝胶法、水/溶剂热法、固相反应法(研磨、高温焙烧、熔融)、火焰热解法等; 负载催化剂的制备方法主要有浸渍法、沉积-沉淀法、离子交换法、物理/化学气相沉积法、化学嫁接/接枝法等; 催化剂制备过程中主要包括离子吸附/交换、沉淀、老化、过滤(包括洗涤)、蒸发、干燥、焙烧等操作单元。本章从溶液化学、晶体生长、材料表/界面物理化学性质等基础概念入手, 选取最常用的沉淀/共沉淀法、溶胶-凝胶法、浸渍、离子交换法以及沉积-沉淀法等液相化学方法, 试图对催化剂制备过程单元操作步骤中发生的物理、化学变化进行解析, 重点介绍固体催化剂制备的基本原理。
2.2 本体催化剂
2.2.1 沉淀/共沉淀法
2.2.1.1 成核和晶体生长
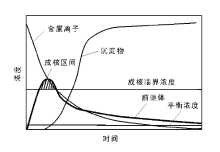
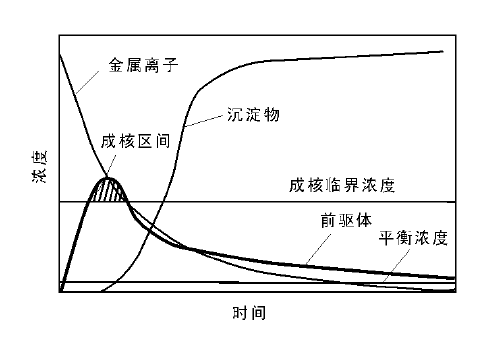
从溶液中获取沉淀物可以通过物理变化(如改变温度、溶剂或直接蒸发)和化学变化两种方式实现, 通常所说的沉淀法是指化学沉淀法。化学沉淀法是通过向溶液中加入酸、碱或络合剂等能与溶液中金属离子发生化学反应生成固态化合物, 并使之从溶液中沉淀出来, 从而获得固体材料的一种方式。溶液中的金属离子首先与沉淀剂反应生成前驱体, 当体系内前驱体达到一定浓度后, 沉淀过程才真正开始, 一般经历成核、晶体生长和粒子聚集等三个阶段(图2-1)。
 | 图 2-1 沉淀过程示意图[6] |
成核和晶体生长是沉淀形成过程中两个重要阶段, 一般同时进行, 严格意义上来讲很难完全分离开。成核阶段, 金属离子与沉淀剂离子结合后先形成最小的基元固态粒子, 也称之为晶体胚胎(crystal embryo), 目前对胚胎的结构认识不清晰, 一般认为它是一个原子、分子甚至离子堆积形成的可以自发生长的团簇[7, 8, 9, 10, 11]。团簇存在一个临界尺寸, 小于临界尺寸的团簇会发生溶解再结晶, 而大于临界尺寸的团簇会作为晶核继续生长, 也就是晶体生长过程。溶液中成核沉淀(假设为球形粒子)的吉布斯自由能变化可以表示为:△ G=
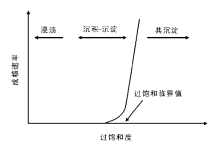
成核速率可以表示为[7, 10, 11, 12, 13]:
 | 图 2-2 成核速率与过饱和度的关系[15] |
晶体生长速率可用公式kg=a(C-Ceq)n表示[15], a是常数, C和Ceq分别是沉淀化合物分子的实际浓度和饱和浓度(溶解度), 在水溶液中多数氢氧化物和碳酸盐化合物的Ceq趋于0, 指数n一般接近1。可见, 成核过程对浓度/过饱和度的依赖是指数关系, 而晶体生长过程则是线性关系, 因此高过饱和度对成核过程的促进作用远大于晶体生长过程。所以通过控制沉淀反应在高过饱和度下进行, 使成核速率远大于晶体生长速率, 容易获得高度分散的小晶粒沉淀物; 反之, 稀溶液中低过饱和度情况下成核速率接近或小于晶体生长速率, 溶液中的金属盐前驱体主要用于晶体生长, 有利于形成大尺寸晶体。利用这一原理, 可以通过控制沉淀化合物分子在溶液中的浓度/过饱和度控制成核和晶体生长的相对速率, 将沉淀过程分成以成核为主和以晶体生长为主的两个阶段, 即所谓成核和晶体生长过程的分离, 这是调控晶体尺寸和形貌均一性的一种重要手段。在粒子尺寸均一的单分散晶体制备过程中, 一般采取的策略是通过提高初始过饱和度完成快速成核, 使体系内晶核数目短时间内达到极大值, 此后由于过饱和度降低, 成核过程速率急剧下降, 成核过程结束, 晶体生长与晶核(小于临界尺寸的晶核)溶解过程将体系维持在一个准平衡状态, 即奥斯瓦尔德熟化过程[6, 11]。
除采用高的初始过饱和度外, 还可以采取预先加入晶种的方式直接实现成核和晶体生长的分离。此时不需要再发生成核过程, 体系会以加入的晶种为晶核直接开始晶体的生长过程。在此情况下, 沉淀速率一般可以用阿伦尼乌斯速率方程来描述, 如拜耳工艺生产氧化铝的过程中, 沉淀速率可以用如下方程描述:-
2.2.1.2 沉淀物结构性质的影响因素
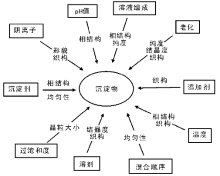
沉淀物的结构和物理化学性质受多种因素影响, 如前驱体金属盐、沉淀剂和溶剂等的选择, 以及温度、溶液的pH值、过饱和度、试剂的混合方式、滴加速率、搅拌速率等操作条件的控制, 如图2-3所示。通过这些条件的改变可以调控沉淀物的结构和性质, 如晶相、化学组成、纯度、粒子大小、表面积和孔结构等。
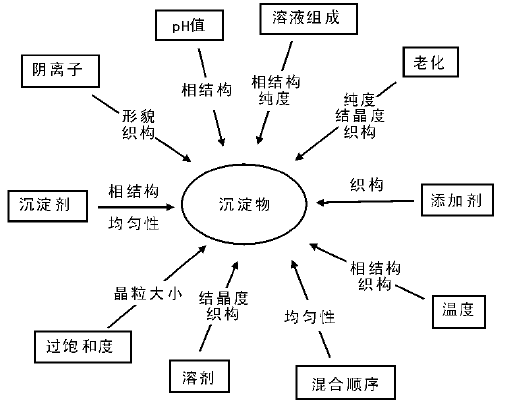 | 图 2-3 沉淀物结构和性质的影响因素[17] |
(1) 原料和溶液组成
原料的选用需要考虑多种因素, 比较重要的是其溶解性, 实际应用中还需要考虑原料是否环境友好、是否易于获取、经济性等。需要指出的是原料对目标产品的结构性质可能造成重要影响, 例如前驱体盐阴离子残留杂质对沉淀物的影响, 因此原料的选择需要结合多方面因素, 综合考虑。通常制备高分散的沉淀物时, 具有高溶解性盐和沉淀剂便于在高过饱和度下操作, 易于获得晶粒尺寸小的沉淀物。一般最常用的金属盐是硝酸盐, 其中比较重要的一个原因是硝酸根离子比较易于通过洗涤和焙烧等后续操作进行清除。氯化物和硫酸盐等也比较常用, 但在某些催化过程中Cl-和S
溶液的组成, 如反应体系中阴离子以及溶液浓度的变化, 是影响沉淀物结构性质(如晶粒尺寸、形貌以及晶相等)的重要因素。如分别以Na2MoO4和(NH4)6Mo7O24(沉淀pH分别为1.65和2.30)为原料采用沉淀法制备MoO3时, (NH4)6Mo7O24沉淀可得到晶粒尺寸更小的MoO3, 且催化剂上四面体钼物种的比例更高[18]; 又如改变溶液中FeCl3和HCl浓度, 控制反应条件可得α -Fe2O3(0.031 5 mol· L-1FeCl3, 0.005 mol· L-1HCl, 100 ℃, 2 周)和β -FeOOH(0.27 mol· L-1FeCl3, 0.01 mol· L-1HCl, 100 ℃, 24 h); 而改变阴离子后, 在含有Fe(NO3)3和Na2SO4的体系中[0.18 mol· L-1Fe(NO3)3, 0.32 mol· L-1Na2SO4, 98 ℃, 2 h]沉淀生成Fe3(OH)5(SO4)2· 2H2O; 在FeCl3和H3PO4体系中(0.003 8 mol· L-1FeCl3, 0.24 mol· L-1H3PO4, 100 ℃, 20 min), 沉淀生成FePO4[19]。上述得到的沉淀在形貌上也有很大差异, 如图2-4所示, 在FeCl3和HCl体系中, 控制反应物浓度和老化时间可以得到球形(图2-4a)和棒状(图2-4b)结构; Fe(NO3)3和Na2SO4体系中短时间内即可得到六角形状Fe3(OH)5(SO4)2· 2H2O沉淀物(图2-4c)。溶液的组成还可能决定沉淀物的晶相组成、表面积、热稳定性等物化性质[20, 21, 22]。CuO/ZnO催化剂的制备过程中, 将含有Cu/Zn不同的溶液[Cu(NO3)2+Zn(NO3)2=1 mol· L-1]加至过量的NaHCO3溶液(1.2 mol· L-1)中进行沉淀, 结果发现, 当Cu/Zn大于85/15时生成类孔雀石相[malachite, Cu2CO3(OH)2], 而随Cu/Zn降低(50/50≤ Cu/Zn≤ 77/23)沉淀物中出现绿铜锌矿相[aurichalcite, (Zn, Cu)5(CO3)2(OH)6], 且沉淀物混合相结构中绿铜锌矿相的比例随Zn含量增加而增大[20]。共沉淀法制备MoO3/ZrO2催化剂过程中发现, (NH4)6Mo7O24的加入(0< Mo/Zr≤ 0.5)使催化剂比表面积增大, 并对介稳的四方相ZrO2向单斜相转变有抑制作用, 500 ℃焙烧后, 单独ZrO2的比表面积只有42 m2· g-1, 当Mo/Zr为0.2时, 催化剂的比表面积最大可达116 m2· g-1[21]。
 | 图 2-4 不同铁盐沉淀物的扫描电镜照片 [19] |
此外, 某些有机物的加入也影响沉淀物和催化剂的结构性质。如在氢氧化铝制备过程中加入能强吸附在沉淀物表面有机物(如丁醇、邻苯二甲酸酐、苯甲酸等)可以改变沉淀物的孔结构, 这是由于这类有机物的吸附取代了吸附在沉淀物表面的水, 可以降低沉淀溶解度, 抑制奥斯瓦尔德熟化过程, 同时有机物较大的分子尺寸有利于抑制沉淀粒子间的聚集[23]。此外, 使用醇类溶剂对沉淀物进行洗涤一般有利于形成介孔结构、增大比表面积, 这是由于醇类取代沉淀物孔内的水后, 其较小的表面张力有利于减缓焙烧过程中孔结构的坍塌[23, 24]。但也有报道发现, 用醇类洗涤Mg(OH)2沉淀物会引起MgO比表面积降低, 这可能是由于醇类与沉淀物表面反应形成烷氧化物, 在焙烧过程中诱导粒子间通过表面缩合反应桥连和聚集[25]。
(2) 操作条件
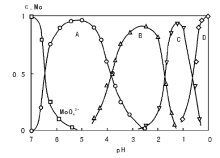
溶液pH值和反应温度是控制沉淀物结构性质的重要参数, 对沉淀物的化学组成、晶相结构、孔分布等有较大影响[27, 28, 29, 30, 31, 32, 33, 34, 35, 36]。首先, 沉淀法中一般通过pH值的调节控制过饱和度, 因此调变pH值是控制沉淀粒子大小和孔结构的常用手段。pH值的变化还可能造成沉淀物相结构的不同, 在氧化铝制备过程中, 较高的pH下(pH> 8)沉淀得到β -Al(OH)3, 而在偏酸性的条件下沉淀得到γ -AlO(OH), 即薄水铝石, 后者焙烧后可得到高比表面积的γ -Al2O3。此外, 金属离子在溶液中的化学状态(配位状态、聚合度等)也依赖于pH值, 比较典型的如V、Mo、W等金属盐随pH值变化而呈现不同的聚合度[27, 28, 29], 图2-5给出了钼物种的化学状态与溶液pH值的关系。从图2-5可以看出, 随pH值从6降至1, 钼物种逐渐由二聚向更高的聚合状态转变, 继续降低pH值钼物种由带负电荷转变为正电荷。因此, 钼酸盐、钨酸盐等化合物的化学组成直接取决于沉淀过程的pH值, 这在钼酸铁、钼酸铋和钨酸铋等混合金属氧化物制备过程中有重要应用, 表2-1列出了pH值和温度对一些钼酸盐和钨酸盐化学结构的影响[30]。催化剂制备过程中, 为了获得结构性能优异的催化剂, pH值的选择需要考虑多方面的因素, 通常结合实验手段进行选择。譬如在多金属离子共沉淀制备催化剂过程中, 由于不同金属离子沉淀的初始pH值差异较大, 一般需要选择在稍高的pH值下进行操作, 但过高的pH值又可能引起某些物相/组分的溶解(与沉淀剂的选择也有关系)。典型的例子如, Cu-Zn-Al催化剂的制备过程中, Al3+、Cu2+和Zn2+开始沉淀的pH值分别约为2.5、3.0和4.5, 实验表明, 控制沉淀pH值为6~7时得到的沉淀物能获得高活性的甲醇合成催化剂[34, 35]; pH值为7时, 沉淀物为类孔雀石相[malachite, (Cu, Zn)CO3(OH)2]结构; 而当pH值< 6时, 沉淀物则为碱式硝酸盐, 这可能是造成催化剂性能差异的原因[36]。从2.2.1.1的介绍可以知道, 晶体生长的动力学对温度变化非常敏感, 但由于不同沉淀过程基元步骤的动力学行为不同, 实际操作中, 很难准确预测温度变化如何影响沉淀物的某个具体性质。例如, 升高沉淀温度通常有利于得到大晶粒尺寸的产物(如在拟薄水铝石和钼酸铁合成中), 但在ZnO合成过程中也观察到升高温度导致晶粒尺寸的减小的现象[33]。
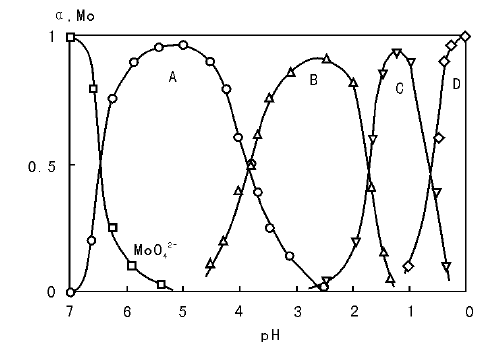 | 图 2-5 钼物种的化学配位状态与pH值的关系[26] |
| 表 2-1 若干钼酸盐和钨酸盐的制备条件[30] |
(C)Mo36
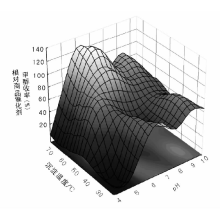
另一方面, 温度和pH值对沉淀物结构性质特别是催化性能的影响是相互关联的, 在特定温度下的最优化(指催化性能相对较好)pH值, 在温度改变后也随之发生变化; 反之亦然。因此, 为获得最佳的催化剂制备条件, 需要通过大量实验逐步对每个变量进行优化。图2-6给出了温度和pH值变化对共沉淀法制备Cu-Zn-Al催化剂甲醇合成活性的影响规律。在沉淀温度为(60~70) ℃和pH值为6~7时, 甲醇收率达到极值, 这是由于在此条件下沉淀生成类孔雀石相碱式碳酸盐, 其结构中铜离子和锌离子同晶取代, 其位置可以相互替换, 使铜和锌之间具有更强的相互作用[37]。
 | 图 2-6 沉淀温度和pH值对Cu-Zn-Al甲醇合成催化剂反应性能的影响 [37] |
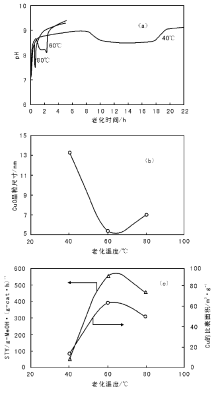
需要特别指出的是, 初次沉淀物一般是非热力学稳定的化合物, 结构中可能含有前驱体金属盐阴离子, 如C
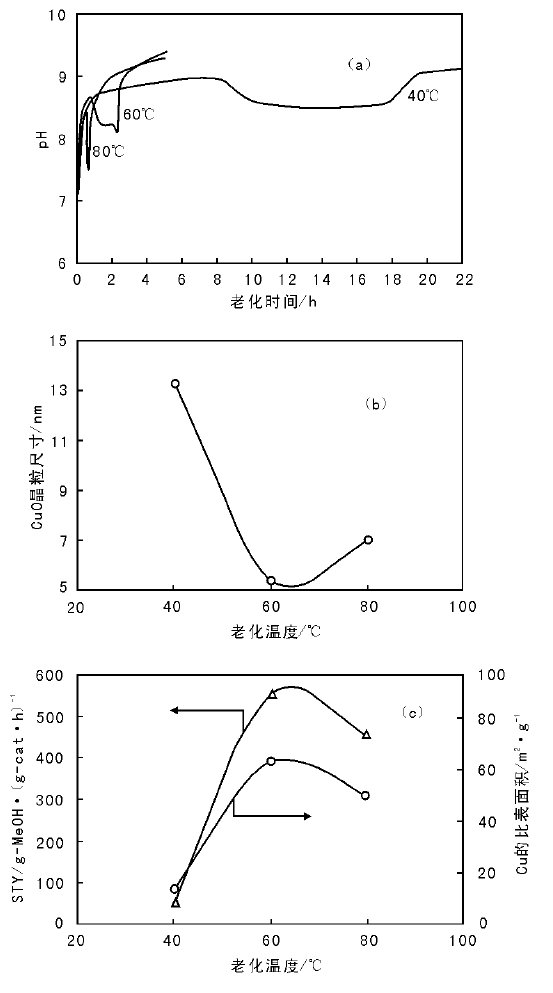 | 图 2-7 pH值随老化时间的变化(a), CuO晶粒尺寸(b) 、Cu的比表面积和反应活性与老化温度(老化5 h)的关系(c)[43] |
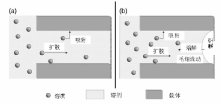
沉淀法制备催化剂一般采用间歇式操作, 溶液的混合方式/顺序需要特别关注。一般有三种混合方式:(1) 将碱液加到盐溶液中; (2) 将盐溶液加入碱液中; (3) 盐和碱溶液同时加入。当将碱溶液加入到盐溶液中时, 随加入量增加pH值逐渐增大, 沉淀的前期过程中由于pH值较低, 产物中可能含有大量盐前驱体的阴离子, 这种情况下需要对沉淀物进行充分老化, 以便将前驱体盐阴离子置换掉。在多金属离子共沉淀时, 由于各金属离子沉淀的pH值不同, 将碱逐渐加入盐中的方式会导致各金属离子在不同pH值时分别沉淀, 造成沉淀物的不均匀分布, 而采用将盐加入碱中的方式可以解决这一问题。上述两种混合方式条件下, 体系的pH值都变化, 可能造成不同pH值下沉淀物结构性质的变化, 因此沉淀物结构均匀性难以保证。碱和盐溶液同时加入的方式能够使体系pH值维持恒定, 一定程度上解决了pH值变化造成的影响。在实际过程中, 碱和盐溶液混合方式的不同可能引起催化剂结构和性能的巨大差异, 如图2-8所示, 在氧化铝制备过程中, 控制溶液最终的pH值为9, 当将1 mol· L-1的Al(NO3)3加入3 mol· L-1的NaOH溶液时得到薄水铝石; 反之, 则得到三羟铝石; 而同时加入时得到纤维状薄水铝石[46]。
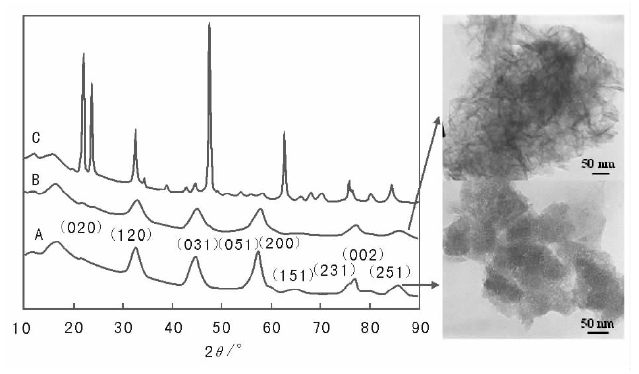 | 图 2-8 混合顺序对氧化铝结构的影响(A)盐和碱同时滴加; (B)盐加入碱中; (C)碱加入盐中[46] |
由此可见, 操作条件对沉淀物结构性质的影响非常复杂, 对各种操作条件的考察大都是在固定其他操作条件不变的情况进行, 针对特定体系内得到的结论。实际上, 特定催化剂的优化制备条件都是经过长期大量的试验摸索出来, 商业催化剂的制造一般严格按照特定操作参数进行。因此, 需要针对催化剂制备的溶液化学开展系统性的基础研究, 理解各种参数对催化剂结构变化的物理化学原理, 实现催化剂结构(关键是本征活性位结构)的定向设计合成。
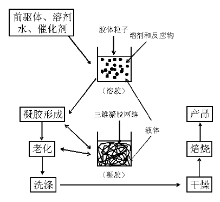
2.2.2 溶胶-凝胶法
溶胶-凝胶法是将溶液中的化合物逐渐转变成溶胶, 然后溶胶粒子交联形成凝胶, 并最终转化成固态干胶或气溶胶(图2-9)。其特征在于:反应体系中的前驱体化合物首先通过水解和缩聚反应形成稳定的胶体溶液[胶体粒子大小(1~100) nm], 然后胶体粒子间进一步通过表面活性官能团间的聚合形成网络多孔结构(孔径一般为亚微米级), 即凝胶; 凝胶网络内包含有溶剂和聚合生成的副产物, 通过洗涤、干燥最终形成干胶或气溶胶[47, 48]。传统上, 凝胶的生成一般是通过控制金属离子或醇盐的水解和聚合反应实现, 近年来还发展了金属盐(如氯化物)醇解法、氯化物与金属醇盐直接聚合反应等非水相合成方法[27, 49]。溶胶-凝胶法大致可分为以下几个操作步骤:(1) 将前驱体转化为活性反应物(活化), 如将醇盐水解成羟基化合物; (2) 活化后的前驱体分子进行聚合反应生成纳米团簇性质的胶体溶液(溶胶); (3) 溶胶转化成凝胶; (4) 老化; (5) 洗涤; (6) 干燥; (7) 热处理/焙烧[47, 48, 49, 50]。在上述步骤中有多个可控的参数, 如前驱体的选择、溶剂、温度、水解和聚合催化剂的选择、老化和干燥条件等等, 因此该方法在控制催化剂结构、织构、组成均一性等方面有独特的优势。
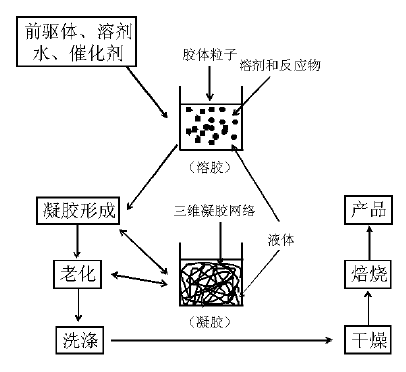 | 图 2-9 溶胶-凝胶法过程示意图 |
2.2.2.1 凝胶的制备
前驱体分子的选择决定了制备过程中可能发生的化学反应, 当前驱体分子不能直接进行聚合反应时, 需要先将其转化成易于聚合的活性分子, 称为活化过程。例如, 当以醇盐化合物作为前驱体时, 一般需要先将醇盐化合物进行初步水解, 使之转化为含有羟基的化合物。但这个步骤不是必须的, 例如通过金属氯化物与醇或醚反应, 这类无水溶胶-凝胶法中不需要这个步骤。金属盐进入水中后在水的溶剂化作用下与阴离子解离并形成金属离子配合物[M(H2O)n]Z+, 水通过氧原子与金属配位, 水相当于路易斯碱, 而金属离子相当于路易斯酸, 金属离子接受氧提供的电子使氧带部分正电荷活化了O— H键(即极化), 并导致其断裂:[M(H2O)n]Z++hH2O=[M(OH)h(H2O)n-h](Z-h)++hH3O+, 即水解过程, h的大小取决于金属离子极化能力的强弱(与金属离子浓度和溶液pH值也有关系)[27, 51]。显然, 当向溶液中加入碱时, 金属离子水解程度加大, 此外向溶液中加入能接受质子的有机物(如环氧丙烷)也可以促进水解。如由FeCl3合成Fe2O3的过程中, 环氧丙烷接受质子后发生开环反应, 反应消耗质子和FeCl3中的Cl-, 给水解反应提供OH-[52]。这种加入环氧化物的方式可以更好地控制金属离子的水解和后续胶体及凝胶的形成过程。有些高价金属离子在水溶液中以阴离子状态存在, 如Si、Mo、V、W等{V
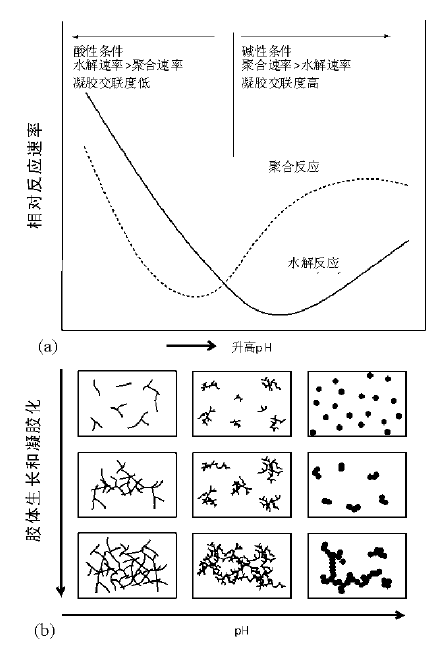
金属醇盐化合物是常用的前驱体, 这类化合物的活化也是通过水解形成氢氧化物完成的, 其水解活性与化合物分子中金属所带的电荷(δ )有关, 取决于金属的电负性。在正硅酸乙酯Si(OC2H5)4中, δ =+0.32, 因此其水解活性较低, 可以通过加入催化剂(如酸)提高其水解活性, 而Ti(δ =+0.63)和Zr(δ =+0.74)等荷电高的金属醇盐化合物具有高的水解活性, 需要通过水的浓度、反应温度或加入其他有机配体控制水解速率和水解反应进行的程度[49]。正硅酸乙酯在酸或碱催化下水解反应是SN2亲核取代过程, 反应速率在pH值=7附近达到极小值。除pH值外, 反应速率还受烷氧官能团大小和水解程度(水解产物分子中羟基与烷氧基的比例)的影响[54]。
反应物前驱体(金属盐或金属醇盐)经水解活化后生成羟基/多羟基化合物, 分子间发生缩合反应生成聚合物种, 形成胶体溶液(溶胶)。这个过程通常和活化过程同时进行, 视前驱体分子的水解程度以及反应温度和加入催化剂(酸或碱)的不同, 多羟基化合物分子可以通过脱水反应聚合, 也可能通过脱醇类聚合[49]。初步聚合反应生成包含几个金属原子的单聚或多聚体, 通常以由金属为中心的多面体通过共用边角棱等方式组成的原子簇形式存在, 随着水解和聚合程度增加, 原子簇初级结构继续聚合长大, 形成纳米胶体粒子。活化和聚合过程都受pH值影响, 以硅酸酯前驱体为例, Si(OR)4-n(OH)n在低pH值时(pH< 4)水解速率大于聚合速率, 因此低pH值时初始聚合物一般多为链式结构, 反应后期逐步交联形成多孔网络; 而高pH值下初始反应即生成交联的网状聚合物, 容易生成大尺寸聚集体(图2-10)[49, 55, 56]。此外, 聚合速率随硅酸酯水解程度增加而增大, 即Si(OR)4-n(OH)n中n值越高, 聚合速率越快。
对无水体系中的溶胶-凝胶过程, 基于灵活的反应体系设计, 可以存在多种活化和聚合的方式, 如金属醇盐与金属氯化物直接反应脱除氯代烷烃(M-Cl+M-OR=M-O-M+R-Cl); 金属氯化物与乙醚或丙酮反应利用有机物中的氧进行桥连聚合(M-Cl+R-O-R=M-OR+R-Cl); 金属醇盐与金属羧酸盐反应脱除酯类化合物(M-OR+M-OCOR’ =M-O-M+R-OCOR’ )[57, 58, 59]。与传统溶胶-凝胶法相比, 无水体系的溶胶-凝胶法可以利用更廉价金属氯化物等为原料, 通过氧桥连机制和反应介质的改变可以更好的控制反应动力学过程, 从而控制聚合程度, 调变材料的结构、均一性、织构和表面性质等[60]。
上述形成的溶胶体系经过“ 胶化” 过程转化成凝胶。胶化是一个将初级胶体粒子缩聚成无限扩展的胶体网络的过程, 本质上是初级胶粒表面官能团间通过氧桥连反应的进一步聚合。由溶液状态转变成弹性胶体的这段时间定义为“ 胶化时间” (gel time), 胶化时间通常由实验手段测得[50]。经过这个过程, 流动态的液体转变成凝胶网络, 由于其空隙间包杂溶剂, 因而是可承受一定压力的“ 弹性” 体[56]。胶化是溶胶-凝胶过程中比较关键的一步, 溶液温度、pH值、水含量和催化剂(对水解和聚合过程起催化作用)是关键参数, 这些条件控制不当可能造成胶体粒子的快速沉淀, 引发固液分离。通常凝胶的形成需要控制反应在温和条件下进行, 如对pH值的调节, 可以通过碱溶液的缓慢加入或使用尿素缓慢释放OH- [61]; 使用环氧化物作为质子捕获剂也可以有效控制体系的pH值[62]。此外, 加入络合试剂(如羧酸、β -二酮等)与金属离子形成配合物也可降低水解反应速率; 调节胶体表面羟基浓度也能对“ 胶化” 过程进行有效控制[63]。凝胶的结构很大程度上取决于金属— 氧键(M— O)的离子性与共价性, 同时受控于活化和聚合反应速率(图2-10)。如Si— O键的共价性约为50%, 与其他金属(Al、Ti和Zr等)相比, Si— O— Si键的键角变化可以更大, 更易于生成链式或开放的网络结构凝胶[56]。
2.2.2.2 老化、洗涤和干燥
凝胶生成后通常需要进行老化处理, 在这一阶段胶体粒子间进一步聚合, 同时胶体发生“ 脱溶剂收缩” , 并可能伴随晶粒长大和晶相结构变化[50]。随老化时间延长, 凝胶中的胶体粒子间通过表面官能团的脱除进一步聚合, 凝胶体系交联度持续增大。核磁研究表明, 以硅酸酯为原料溶胶-凝胶法制备SiO2的过程中, 凝胶形成后反应体系内Si-OH的数量继续随时间延长而减少, 产生新的化学键, 形成交联结构[64]。随聚合程度进一步增加, 可能导致凝胶孔结构的收缩, 孔内的溶剂被挤压出来, 这个过程通常称为“ 脱溶剂收缩” 。实验结果表明, 在水溶液中SiO2凝胶的“ 收缩” 速率在等电点时(pH=2.0)最小, 此时聚合反应速率接近最低, 说明胶体粒子的聚合是引起“ 收缩” 原因[65]。同时, 胶粒在熟化作用下发生溶解再结晶过程, 晶粒尺寸长大, 还可能由无定形或亚稳相向稳定的晶相结构转变[65, 66, 67, 68]。老化过程中的温度、时间、pH值以及溶剂等都可能影响凝胶孔结构和比表面积的变化, 温度升高、老化时间延长均可以促进胶体粒子间的聚合, pH值、溶剂性质影响胶体粒子的溶解再结晶过程。采用合适的溶剂对凝胶进行洗涤或将凝胶浸入到其他溶剂中, 可以将残留在孔内的反应物和溶剂置换掉。如果溶剂置换在老化步骤前进行, 可以通过溶剂性质对老化过程中凝胶结构的变化进行调控; 如果老化完成后再进行溶剂置换, 溶剂的性质可以影响凝胶在干燥过程中的结构变化。
干燥过程也称为脱溶剂过程, 是通过将凝胶结构中包杂的液体脱除, 将凝胶转化成多孔的固体材料的步骤。通常采用溶剂挥发和超临界干燥两种方式, 得到的相应材料分别称为干胶和气溶胶。溶剂挥发一般分为三个步骤:(1) 溶剂的挥发伴随毛细作用力下的凝胶网络收缩; (2) 伴随凝胶网络收缩和堆积密度增大其机械强度增加, 凝胶收缩达到“ 临界点” , 孔内的溶剂在毛细凝聚梯度作用下开始流到外表面; (3) 剩余溶剂的挥发。步骤(1)中凝胶网络/孔的收缩是由于毛细作用力下孔内外压差造成的, 压力作用的大小可用杨-拉普拉斯公式来表示:△ P=2γ cosθ /r(γ 是液体的表面张力, θ 是液固接触角的大小, r是孔半径), 如当大小为1 nm的孔填充溶剂为水时, 压差可以达到1.5× 108 Pa[67]。步骤(2)开始前大部分溶剂从凝胶体系中挥发出来, 体系达到“ 半干” 状态, 此后溶剂挥发速率降低[65, 66, 67, 68]。通过采用低表面张力或者与固体接触角大的溶剂可以降低毛细作用力引起的孔结构收缩/塌陷, 使孔结构得到保持。在SiO2合成中, 采用乙酸洗涤, 置换掉凝胶孔结构中的水, 可以减小干燥过程对孔结构的破坏, 这是由于乙酸的表面张力仅为水的1/3[65]。
超临界干燥法是一种可以将凝胶结构完整转化为气溶胶的有效手段, 超临界流体不会浸润凝胶, 因此不会由于毛细凝聚力对凝胶孔结构造成破坏。常见的超临界干燥方式有两种:(1) 将凝胶在加热加压作用下使溶剂达到超临界状态, 然后再将超临界溶剂缓慢释放出(超临界溶剂释放法); (2) 将超临界流体(如超临界CO2)流过凝胶, 在此过程中凝胶内的溶剂被超临界流体萃取出来(超临界萃取法)。超临界干燥可以获得具有丰富孔结构的大比表面积固体材料, 且材料一般有低密度、织构稳定性好等特点[69]。由于水在超临界状态下(647 K, 22.1 MPa)通常会将凝胶溶解, 对固体的结构造成破坏, 因此一般采用醇或醚类作为超临界介质[67]。超临界干燥法一般在高温下进行, 温度对所形成的气溶胶的性质有较大影响。在第一种操作方式下, 应严格控制压力释放的速率, 防止由于压力释放过快, 溶剂来不及从孔内流出就剧烈膨胀而对孔结构造成破坏。超临界萃取法一般使用超临界CO2为溶剂, 在温度(310~318) K和压力(8~30) MPa半连续状态下进行, 萃取前通常会先将凝胶中残存的水用醇类或CO2交换掉, 这样可以增加溶剂在超临界CO2中的溶解性。压力和温度仍然是影响催化剂织构性质的主要参数, 通常温度升高时有利于获取大孔径的产物。
2.3 负载型催化剂
负载型催化剂由载体和分散在载体表面的催化活性组分组成, 一般采用大比表面积的载体来提高活性组分的分散度和抗烧结/聚集能力。载体可以是惰性的(即载体对催化反应没有活性或载体对活性组分的性能无明显影响), 但多数情况下载体的存在会影响活性组分的性质和催化性能, 或者载体本身也具有一定催化活性, 构成双/多功能催化剂[70, 71, 72]。将活性组分通过简单易操作均匀地分散在载体表面是制备高效负载型催化剂的关键, 常用方法有浸渍法、沉积-沉淀法、离子交换法、固载化和嫁接法、物理和化学气相沉积法等, 其中, 浸渍法、沉积-沉淀法和离子交换法是采用将活性组分先分散在溶液中再转移到载体上的方式。因此, 固液界面间的物理和化学相互作用机制是控制活性组分分散度和结构状态的关键。从微观角度讲, 当将载体放置到溶液中时, 可能发生溶液对载体表面和孔结构的浸润、离子交换、载体的部分溶解和形成新表面物种等诸多过程, 因此, 上述对活性组分负载方法的分类只是从宏观角度进行的描述, 体现了制备过程中对活性组分分散起主要作用的物理、化学作用机制[70]。
2.3.1 金属离子在载体表面的吸附
金属离子与载体表面的作用对活性组分的分散至关重要。活性金属组分前驱体溶液浓度较低时(如贵金属), 金属离子与载体的作用是发生表面吸附的主要驱动力; 浓度较高时, 金属离子与载体之间的相互作用对随后干燥、焙烧过程中活性组分在载体表面的晶种形成和结晶过程也有重要影响[70, 71, 72]。为提高金属含量和分散度, 通常也会采用多步浸渍的方法, 后续浸渍的物种在浸渍和干燥过程中会以前一步浸渍形成的晶种为中心进行聚集[73]。金属离子在载体表面的吸附方式主要有两种, 即静电吸附和表面化学反应, 其中静电吸附应用较为广泛。
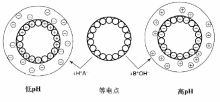
溶液中带有正/负电荷的金属离子通过静电吸引与载体表面发生作用, 金属离子从溶液中转移到载体表面, 同时载体表面的平衡离子转移到溶液中, 这一过程一般称为离子交换。其原理是利用溶液中离子与载体之间更强的静电作用, 或溶液中高浓度的金属离子与载体本身的平衡离子之间的吸附平衡, 将溶液中的催化活性组分前驱体转移到载体上, 根据这一原理发展的催化剂负载方法称为离子交换法。根据离子交换作用的差异, 一般可以将载体分为两类, 即本身具有平衡离子的载体(如交换树脂、粘土、分子筛等)和两性载体, 前一类载体本身具有平衡离子, 受平衡离子荷电性质的控制只能交换阴离子(如水滑石)或阳离子(如分子筛)中的一种; 而两性载体根据金属中心电负性的不同, 可能带正电荷或负电荷, 与溶液的pH有关(图2-11), 因此可以通过控制溶液pH值改变载体的Zeta电势, 使其有利于阴离子或阳离子的吸附[71]。
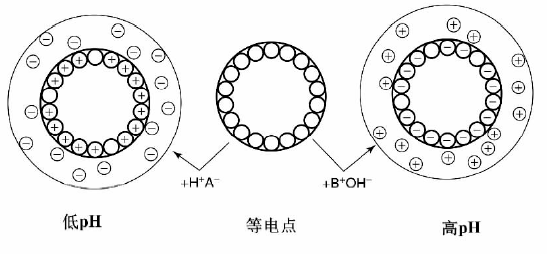 | 图 2-11 两性载体表面荷电示意图 |
两性载体分散于水溶液后, 其表面基本被羟基覆盖, 可以表示为S-OH(S表示Al、Si、Ti、Fe等元素), 根据载体本身性质的不同, S-OH可能表现出Brö nsted酸或碱的性质:S-OH=S-O-+H+, S-OH+H+=S-O
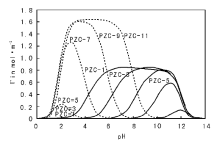
一般PZC值较低的载体在水溶液中表面带负电, 常用于阳离子的吸附, 反之PZC值较高的载体常用于阴离子的吸附, 而PZC值居中(PZC值在4~9)的载体可以通过对pH值的控制改变表面带电类型, 即可用于阴离子吸附也可用于阳离子吸附。因此需要根据载体PZC值的不同, 同时考虑溶液的pH值来选择合适的活性组分金属盐, 以实现载体对金属离子的有效吸附。以常见的Pt盐为例, 图2-12给出了阴离子型Pt盐(如Na2PtCl6)和阳离子型Pt盐[如Pt(NH3)4(NO3)2], 在不同PZC值载体上的理论模拟吸附量随溶液pH值的变化。从图2-12可以看出, 对PZC值较高的载体(如Al2O3)选取阴离子型Pt盐作为前驱体可以有效提高Pt在载体上的吸附量, 反之对低PZC值的载体(如SiO2)应该选取阳离子型Pt盐为前驱体[77]。此外, 特别需要注意的是此处所述的溶液pH值是指将载体置入溶液后的最终pH值, 由于载体加入溶液中后, 表面质子化/去质子化过程会消耗溶液中的H+/OH-, 造成pH升高/降低(这种现象称为载体的“ pH缓冲效应” ), 为使载体表面充分带电, 所需的溶液初始pH值需要远大于或小于PZC值。如对某Al2O3载体, 假设其比表面积为200 m2· g-1, PZC=8.5, 表面羟基浓度为8 OH· nm-2, 若将1 g该载体(MOH=1 g× 200 m2· g-1× 1018 nm2· m-2× 8 OH· nm-2÷ 6.02× 1023 mol-1= 2 660× 10-6 mol)置于1 mL pH值为3的溶液(
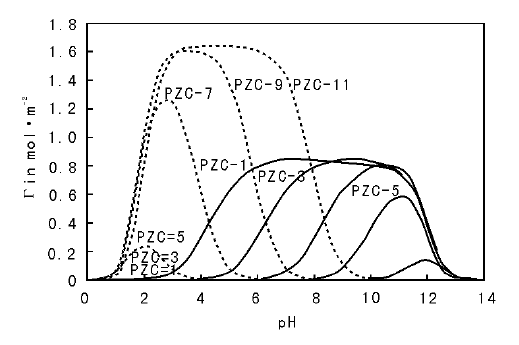 | 图 2-12 不同PZC值载体的理论模拟吸附量随溶液pH值的变化[77] 虚线表示对阴离子的吸附量, 实线表示对阳离子的吸附量 |
表2-2为常见载体的PZC值(范围)[70, 71, 72, 74, 79]。
| 表 2-2 常见载体的PZC值(范围) |
在实际制备过程中, 如果采用自行开发的新型材料作为载体, 或当材料结构、组成等与常见载体区别较大时, 载体的PZC值需自行测定, 常用的方法有电位滴定法、电泳法、粉末加入法(powder addition)以及“ 质量滴定法” (mass titration)等[74, 80]。 “ 质量滴定法” 是一种经典的测定方法, 这种方法是将载体陆续加入到一定pH值的酸(或碱)溶液中, 由于载体表面的质子化(或去质子化)作用, 溶液的pH值会升高(或降低), 随着载体的不断加入, pH值逐渐趋于平稳, 此时的pH值即为载体的PZC值[81]。粉末加入法是将等质量的固体粉末加入到一系列离子强度相同但pH值不同的溶液中, 一定时间后(一般为24 h)测量溶液的pH值变化(△ pH值), 以△ pH值对溶液的初始pH值作图, △ pH值=0的点即为固体的PZC值[82]。这两种方法简便易操作, 但对于比表面积低的载体由于其单位质量的表面OH量小, 需要加入的载体量很大, 导致体系成为浆态状, 很难用常规方法监测pH值。针对这种情况, Jaehyeon Park等[83]专门开发了一种可用于测量准固体(semisolid)的矛尖(spear-tip)电极。
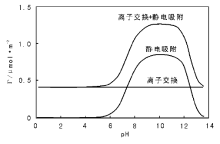
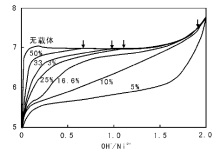
相对其他负载催化剂的制备方法, 这种靠载体与金属离子间的静电吸附方式得到的催化剂, 更有利于实现活性组分的高分散, 在负载贵金属催化剂制备中优势明显。需要说明的是, 有些参考书和文献中对离子交换和静电吸附现象进行了区分。典型的例子是, 以分子筛为载体采用离子交换法制备催化剂的研究中, 发现有些分子筛样品(特别是高Si/Al分子筛)存在“ 过交换” (overexchanged)现象, 即分子筛对阳离子的交换容量大于分子筛中Al的含量[84, 85]。此时对应分子筛中Al含量的交换部分称为离子交换, 而超过Al含量的交换部分称为静电吸附, 两者的区别在于分子筛的理论离子交换量不受pH值影响, 而过交换量/静电吸附与溶液的pH值有关。图2-13给出了ZSM-5分子筛(Si/Al≈ 100)理论离子吸附量、静电吸附量和过交换量(两者之和)与pH值的关系[70, 85]。分子筛载体上的这种过交换现象是由于载体表面除固有的Al-(OH)-Si桥羟基质子交换位(对应于离子交换)外, 还有大量的Si-OH(特别是高Si/Al分子筛), 在高pH值下, 其对阳离子有吸附作用[86]。
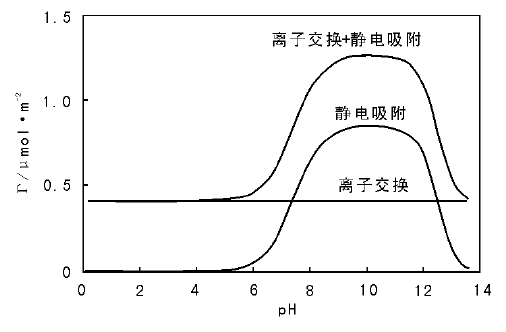 | 图 2-13 分子筛离子交换和静电吸附量随pH值的变化(过交换现象)[85] |
静电作用可以认为是一种物理作用方式, 除静电作用外, 金属离子还可能与载体表面发生化学反应。与单纯静电吸附不同, 金属离子与载体表面官能团通过配位作用, 发生熵驱动的化学反应将金属离子“ 嫁接” 在载体表面, 这个过程涉及到了化学键的断裂和生成, 多发生在浸渍温度较高的情况下或者干燥过程中, 根据化学键强度不同, 该过程也可能是可逆的。如图2-14所示, PtC
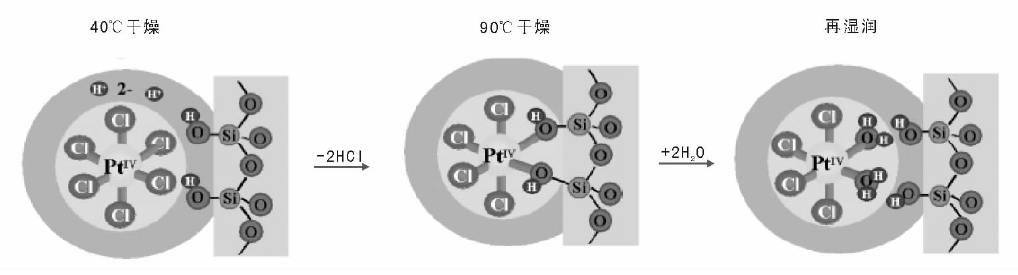 | 图 2-14 二氧化硅载体表面Pt物种的转化示意图[87] |
2.3.2 浸渍法
浸渍法是指将含有活性组分的溶液与多孔载体接触, 活性组分吸附在载体上实现活性组分从溶液向载体表面转移的一种方式。浸渍法通常分为干法浸渍和湿法浸渍。干法浸渍是指控制浸渍溶液的体积与载体孔体积相等, 使溶液刚好完全填充进载体孔内, 又称等体积浸渍法或孔填充法。湿法浸渍是浸渍液体积大于载体孔体积, 液体不能全部进入载体孔内; 当需要负载多种活性组分时, 可将活性组分前驱体溶解后, 在一次浸渍操作中完成负载, 溶液中的活性组分在载体表面竞争吸附, 称之为“ 共浸渍法” (co-impregnation or simultaneous impregnation); 也可以对不同活性组分分次浸渍, 称为顺次浸渍法(successive or sequential impregnation)。负载多组分催化剂浸渍方式和各组分浸渍顺序的选择可能对活性组分结构和催化剂的性能起重要作用。浸渍液所用的溶剂一般为去离子水, 但当载体表面憎水/亲油或需要避免载体水解时, 则选用有机溶剂[89, 90]。
浸渍法制备催化剂一般包括三个主要步骤:(1) 将载体与浸渍液接触; (2) 干燥; (3) 焙烧和催化剂活化。其中前两步对活性组分在载体上的空间分布影响较大, 溶液中的活性组分前驱体与载体间作用强弱和固液接触过程中的传质效率是决定因素。活性组分在载体上的吸附可用Langmuir方程来描述:α s=α m
2.3.2.1 浸渍液与载体的相互作用
浸渍前载体通常需要通过干燥处理以脱除吸附在孔内的水分, 当载体置入液体后, 靠毛细力将浸渍液吸入孔内, 此时封在孔内的空气被压缩, 当毛细凝聚力与孔内气体压力达到平衡时, 液体不能再进入孔内。当载体孔径比较小时, 毛细凝聚力远大于封在孔内的空气压力, 气体可能被溶解或通过孔径更大的联通孔溢出[92, 93]。此外, 在浸渍前将载体进行抽真空处理或者置入NH3气中, 也是排除孔内气体压力的常用方法[89, 90, 94]。载体与浸渍液接触后, 气/固界面转变为液/固界面, 这个过程一般是放热, 如果活性组分前驱体的溶解度随温度升高降低, 或者温度升高导致其他不利于活性组分吸附的反应发生, 都可能影响活性组分的分散, 这种情况下需要避免采用干法/等体积浸渍(等体积浸渍使用的溶液量小, 不利于热量消散)。将载体提前暴露在水蒸汽中, 在载体表面形成一层水性膜可以部分避免上述情况的发生[89, 90]。
载体与浸渍液接触过程中, 影响活性组分前驱体在载体上落位和分布的主要因素有:(1) 溶液中活性组分前驱体向载体表面的扩散; (2) 活性组分前驱体在载体表面的吸附。在等体积浸渍法中, 溶液进入孔内的过程也可能对活性组分的落位造成很大影响, 当溶液内部的传质过程和吸附过程为快速步骤时, 液体沿孔轴向的推进速率造成活性组分不均匀分布[95]。以上所述物理化学作用是产生所谓蛋壳型、蛋白型、蛋黄型以及空间均匀分布结构催化剂的主要原因, 这些类型的催化剂结构在实际应用中各有优势。如蛋壳型催化剂广泛应用于受扩散限制的反应中; 蛋白型催化剂可以克服催化剂使用中由于磨损导致活性组分脱落, 当原料气含有导致催化剂中毒的化合物时也能有效延长催化剂寿命。因此, 利用制备过程中的物理化学机制, 控制活性组分在载体上的空间分布, 可获得所需的催化剂结构[96]。
图2-15是浸渍液与载体接触过程中的吸附和扩散示意图[89]。载体与浸渍液接触后, 表面对活性组分前驱体的吸附造成固液界面附近溶液浓度降低, 外部溶液中的活性组分前驱体通过扩散作用到达固液界面处。溶液进入孔道的时间可以表示为:t=
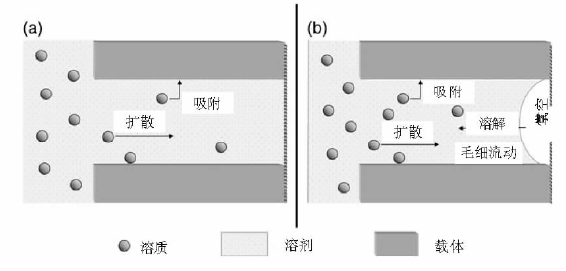 | 图 2-15 湿法浸渍(a)和干法/等体积浸渍(b)过程中溶液和载体的作用示意图[89] |
2.3.2.2 干 燥
浸渍液与载体接触并进入到载体孔内后, 部分活性组分前驱体吸附在载体表面, 但仍可能有部分活性金属离子未能吸附, 特别是高负载量或载体对活性组分吸附较弱时, 活性组分大部分停留在溶液中; 干燥过程中, 载体孔内的浸渍液在毛细流动和扩散作用下发生迁移, 同时活性组分在吸附/脱附过程中进行重新分散[102, 103]。因此需要通过干燥步骤将溶剂挥发, 使活性组分形成理想分布和高分散状态。
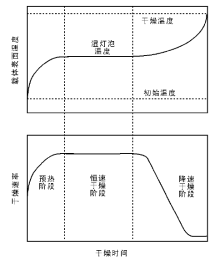
图2-16为载体表面温度和干燥速率随干燥时间的变化示意图[96]。干燥过程一般分为三个阶段:(1) 预热; (2) 恒速干燥; (3) 减速干燥[104]。在预热阶段, 载体逐渐被加热, 溶剂蒸发主要发生在载体外表面, 随着温度逐渐升高, 溶剂蒸发速率也逐渐加快。恒定速率阶段, 随着表面溶剂的挥发, 孔道内的液体流出孔道以维持载体表面润湿, 溶剂挥发速率达到恒定阶段。此时, 溶剂挥发速率取决于热量向载体表面传递的速率, 载体表面温度保持恒定, 形象的称为“ 湿灯泡温度” (wet bulb temperature)[96]。在前两个阶段, 液体呈连续相状态, 载体孔内的液体主要通过毛细流动向外表面扩散, 溶解在液体中的活性组分离子通过对流和扩散进行传输。随着干燥时间的推移, 孔内液体的流动不足以补充外表面液体的挥发, 外表面出现干燥的区域, 溶剂挥发速率放缓(减速干燥阶段)。此时, 外表面温度开始升高, 并逐渐向载体颗粒中心区域辐射, 孔道内的毛细流动减缓, 孔道内的液体开始直接气化并扩散出孔道, 溶剂蒸汽的对流逐渐成为溶剂的主要传输方式, 并成为制约溶剂挥发速率的主要因素[92, 105]。
 | 图 2-16 载体表面温度和干燥速率随干燥时间的变化示意图[96] |
干燥过程中载体上(表面和孔内)液体的传输和扩散对活性组分的再分散有较大的影响。在其他条件已定(载体孔径、孔体积、溶液黏度和表面张力等)的情况下, 干燥温度和采取的干燥方式是影响活性组分再分散的主要因素。干燥温度和干燥方式主要影响溶剂挥发速率, 对溶剂挥发速率假定两种边界情况:快速干燥和慢速干燥。快速干燥一般温度高, 速率快, 溶剂的气化和挥发是主要传输方式, 毛细流动基本可以忽略, 溶剂由外向内逐渐挥发, 不存在恒速干燥阶段; 慢速干燥情况下, 恒速干燥阶段的时间长, 液体运动方式以毛细流动为主。在活性组分弱吸附的情况下, 快速干燥时没有毛细流动造成的活性组分随溶液向载体颗粒表面富集, 有利于形成活性组分均匀分布的催化剂; 慢速干燥时, 如果浸渍液在减速蒸发阶段产生过饱和现象, 活性组分会逐渐沉积, 形成均匀分布催化剂; 如果沉积发生在恒速蒸发阶段, 活性组分大量沉积在载体颗粒外表面, 将形成蛋壳型催化剂[91]。但在活性组分强吸附的情况下, 干燥过程对活性组分的再分散影响较小, 活性组分分布主要取决于浸渍液与载体接触过程中的吸附和扩散行为[106]。
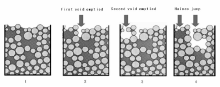
在真实体系中, 液体在干燥过程中的流动和扩散行为更加复杂, 如大的孔穴通过狭窄孔道与外界连接时, 孔穴内的液体可以快速转移到载体表面已经干燥的小孔结构内(Haines Jump), 溶解在液体中的活性组分随之迁移(如图2-17所示)[96]。此外, 伴随溶剂挥发以及溶液浓度变化和活性组分在载体表面的吸附-溶解-再吸附, 同时受传质、传热限制, 将会出现载体颗粒不同区域溶液的浓度和温度差。上述诸因素导致载体不同区域活性组分的聚集度、成核数量等差异较大, 活性组分粒子大小不一[104, 107]。因此, 实际应用中很难对干燥过程中活性组分在载体上的再分散进行定量模拟, 这也就是为什么干燥条件的选择多基于实验验证而非模型研究[92]。除干燥速率外, 干燥方式的选择也很重要, 如微波加热和冷冻干燥等。微波干燥有助于样品整体均匀受热, 与常规烘箱干燥法相比, 微波干燥后, Ni物种在Al2O3上的空间分布更均匀, 催化剂在异辛烷重整反应中失活速率变缓[108]。冷冻干燥中, 水分在冰点温度以下挥发(低压), 不发生液相流动, 保持了活性组分干燥前在载体上的空间分布[109]。
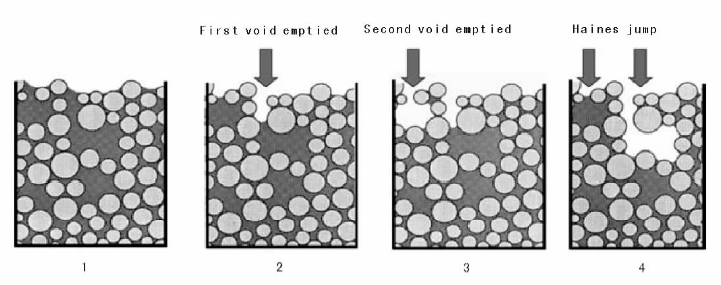 | 图 2-17 多孔材料干燥过程中的Haines跳跃现象 [96] |
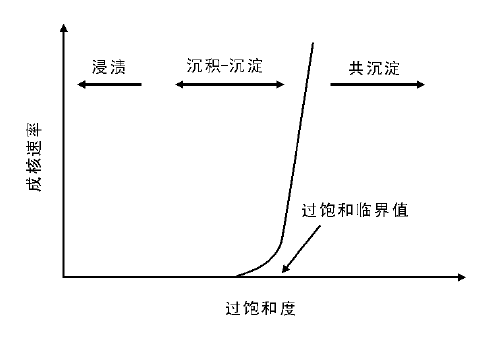
2.3.3 沉积-沉淀法
沉积-沉淀法通过向活性组分溶液中添加沉淀剂, 将高溶解度的活性组分转变为低溶解度化合物, 使之沉淀在悬浮于溶液中的载体表面, 也是制备负载型催化剂的常用方法。与浸渍法相比, 沉积-沉淀法可制备出粒子尺寸小且分布均匀的催化剂, 更适合于制备高金属负载量的催化剂[110, 111, 112]。该方法重复性好, 制备的催化剂载体与活性组分(或共沉积活性组分之间)通常形成金属-载体强相互作用。沉积-沉淀过程还被用来修饰载体, 改善载体的物理结构和化学性质[110, 111, 112, 113, 114]。近年来该方法也用于制备贵金属催化剂, 特别在小尺寸金属粒子的Au/CeO2和Au/TiO2制备中显示出独特的优势[115, 116]。沉积-沉淀方式有多种, 最常用的是通过加入碱溶液提高pH值或加入酸溶液降低pH值[110, 111, 112], 此外改变活性组分化学价态(如还原沉淀)[117]或改变配体的络合状态[118]也是常用的手段。
2.3.3.1 活性组分沉积
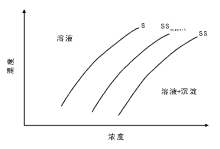
采用沉积-沉淀法制备高分散催化剂的关键是将活性组分选择性沉淀到载体表面, 防止成核和沉淀发生在溶液中。为实现活性组分在载体表面的选择性沉积, 一般需要满足下列条件:(1) 控制活性组分成核浓度, 避免其在溶液中聚集和沉淀; (2) 活性组分与载体表面存在相互作用, 诱导定向沉积。沉淀过程的成核理论(成核吉布斯自由能变化), 同样适用于沉积-沉淀过程。如果将待沉淀物做半球处理, 考虑载体与沉淀物间的界面能, 则沉淀过程的自由能变为:△ G=
 | 图 2-18 载体表面的固液平衡相图 S溶解度曲线, SSsupport载体存在时的过溶度曲线, SS过溶度曲线 |
实现活性组分在载体表面选择性沉积的关键是控制溶液中的沉淀物分子的浓度, 防止局部浓度高于过溶度。以碱溶液加入法为例, 采用逐渐滴加/注射碱溶液操作时, 可能会由于传质受限使得碱溶液局部浓度过高, 局部沉淀物分子浓度瞬时大于过溶度, 导致溶液中快速成核和沉淀, 因此, 采用滴加/注射操作方式时, 必须采用剧烈搅拌以降低溶液的浓度梯度。为解决这一问题, 可以将沉淀剂和溶液的混合与沉淀反应分离开来, 使沉淀过程在均匀溶液中发生; 采取低温混合控制沉淀生成速率, 或采用尿素分解法控制pH值等, 都是常用的有效手段。此外, 采用细小的粉末状载体可以有效增加载体与溶液的接触, 促进传质, 对活性组分在载体表面的选择性沉积有利。
沉积-沉淀过程中, 沉淀分子形成的凝结核与悬浮在溶液中的载体之间的相互作用, 是活性组分在载体表面形成致密、均一包覆沉积的必要条件。没有相互作用存在的情况下, 容易形成大晶粒的沉淀物。静电作用是种比较重要的方式, 静电排斥作用会阻碍金属离子或带电荷的胶体粒子(沉淀核)与载体表面的接近, 但仅有静电吸引作用还不足已保证活性组分的均匀沉积, 通常还需要两者间存在化学作用。比如在沉积-沉淀法制备Ni/SiO2催化剂过程中, 成核阶段镍离子通过与SiO2表面羟基作用成键, 形成Si-O-Ni(OH)(OH2)4物种, 这种强化学作用有利于氢氧化镍与SiO2表面反应生成层状镍硅酸盐(phyllosilicate), 经氢气还原后形成约3 nm且高度均匀分散的Ni粒子[121, 122]。当以活性炭为载体时, 经氢气还原或高温惰性气氛下处理的活性炭表面官能团很少, 载体与镍物种间缺乏相互作用, 沉积-沉淀得大于500 nm氢氧化镍粒子, 且与碳载体分离; 而如果活性炭经过1∶ 1的浓硝酸与浓硫酸混合溶液氧化处理, 表面羧基官能团数量增加后, 可以吸附大量镍离子和原子簇作为成核中心, 增强了沉积物与载体的作用, 镍物种沉积后形成与活性炭紧密结合的镍氢氧化物纳米片层状结构(40 nm× 5 nm), 经氢气还原处理后, 镍粒子的粒径约为8 nm[123, 124]。
在提高pH值引发沉积-沉淀的过程中, 可以通过监测有载体存在和没有载体存在两种情况下沉淀过程的pH值变化来验证沉淀与载体间是否存在相互作用。如采用尿素分解法控制Cu(NO3)2溶液的pH值, 沉积-沉淀制备Cu/SiO2催化剂过程中发现, SiO2载体的存在与否对Cu(NO3)2沉淀过程的pH值变化无影响(图2-19), 沉淀生成碱式盐Cu2(OH)3NO3为低密度、大尺寸的薄片层状物, 沉淀物与载体间不存在强相互作用[127]。如果存在强相互作用, 一般沉淀过程发生在较低的pH值下[125]。如图2-20所示, 沉积-沉淀法制备Ni/Al2O3催化剂过程中, 对照试验表明, 体系内不加入Al2O3时, 随NaOH溶液的加入, NiCl2溶液的pH值迅速升高到极大值(约为7), 而后由于沉淀过程的发生消耗NaOH, pH值变化达到平台期; 而加入Al2O3时, 在pH值约为5.5时即发生沉淀(图2-20, 负载质量分数5%)。低金属负载量时, 引发沉淀的pH值远低于无Al2O3存在时, 这是由于Ni2+与悬浮于溶液中的氧化铝表面形成镍-铝水滑石结构化合物; 随镍负载量的增大, 镍含量超出了能与氧化铝表面生成水滑石结构的范围, 生成Ni(OH)2的量增大[126]。以尿素为沉淀剂制备Ni/SiO2催化剂过程中, 也观察到了类似的现象。如图2-21所示, 当体系中只有硝酸镍和尿素时, 沉淀在约2 h后开始发生, pH值约为6.4; 而加入载体后, 沉淀在约0.7 h后即可发生, 此时的pH值约为5.8, 这是由于沉淀生成了不同于常规氢氧化镍的混乱层状结构, 其与SiO2有强相互作用, 可以转变成层状镍硅酸盐化合物[123, 124]。
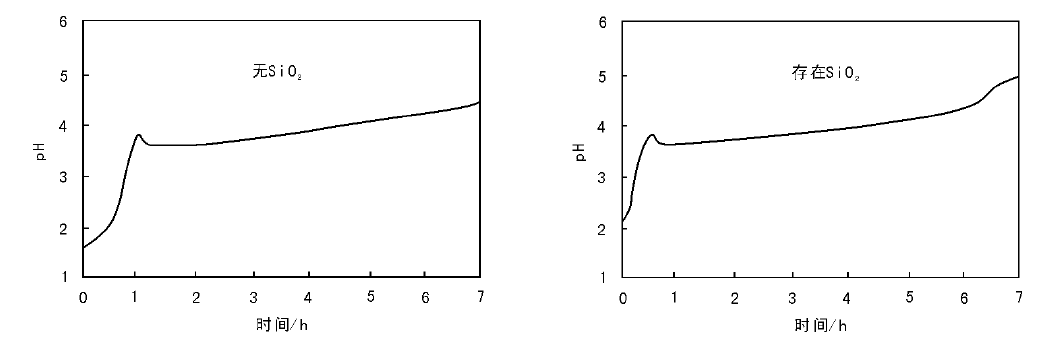 | 图 2-19 SiO2载体对Cu(NO3)2沉淀过程的pH值变化的影响 |
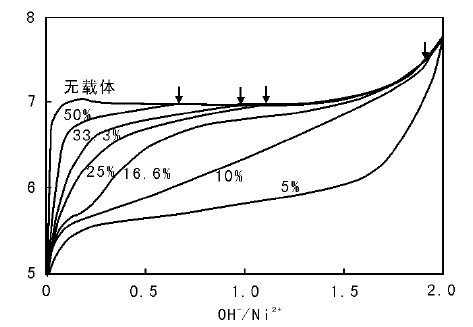 | 图 2-20 不同负载量Ni/Al2O3催化剂沉积-沉淀法制备过程碱滴定曲线[126] 箭头指示与无负载Ni(OH)2曲线交汇点 |
 | 图 2-21 Ni/SiO2催化剂制备过程中体系的pH值变化图[123] 尿素15.3 mmol· L-1, 363 K |
2.3.3.2 沉积物老化[127]
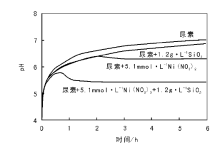
初次沉淀完成后, 沉淀物在载体表面会继续变化, 这其实是一个老化的过程。这类现象比较普遍, 如在Ni/SiO2催化剂制备过程中, 沉淀完成后在70 ℃和90 ℃(pH=7.6)老化有利于Ni物种和载体表面的进一步反应生成镍硅酸盐, 能显著提高Ni物种与载体的相互作用, 具体表现为Ni物种的还原温度由约650 K升高到(700~800) K, 还原后金属Ni的比表面积由(15~20) m2· g-1升高到(60~80) m2· g-1[128]。在初次沉淀物与载体不存在强相互作用的情况下, 老化过程的一个重要作用是可以使初次沉淀物在体系内继续反应, 生成更稳定的物种, 如与其他阴离子结合、与载体表面发生反应等, 这类后续反应有可能生成活性组分与载体强相互作用的物种, 提高活性组分的分散度。沉积-沉淀法制备Cu/SiO2催化剂的过程中, 初次沉淀形成的碱式盐化合物在反应体系内的系列变化即是这类过程的典型代表。
图2-22为Cu(NO3)2沉淀过程的pH值变化。尿素分解法Cu(NO3)2沉淀过程中(没有载体存在时), 初次沉淀过程完成(约6 h)后, 沉淀物在反应体系内老化, 进行到约12 h时, 溶液pH值出现峰值(5.0), 随后在约16 h又出现第二个峰值, (12~16) h的变化对应于碱式硝酸盐转变为碱式碳酸盐的过程, 16 h后碱式碳酸盐转变成Cu(OH)2和CuO。如前所述, 采用尿素分解法将Cu(NO3)2沉积在SiO2上的过程中, 初次沉淀物Cu2(OH)3NO3与载体没有强相互作用, 但SiO2载体的存在会对Cu2(OH)3NO3在老化过程中的变化造成影响。SiO2存在时, 随着尿素继续分解pH值逐渐上升, 达到5.5时又开始下降, 伴随溶液中可溶性铜物种浓度增高, 而后pH值达到平台期, 可溶性铜物种的浓度也基本恒定。随着反应的继续进行, pH值又开始升高, 同时可溶性铜物种的浓度降低。这个过程的物理化学变化细节尚未完全清晰。但初次沉淀物经过上述系列变化最终与SiO2表面反应生成了硅孔雀石结构, 与载体间形成了强相互作用[129, 130]。这种强相互作用的Cu物种具有高的热稳定性, 可以有效防止还原过程中Cu粒子的烧结, 当Cu负载质量分数为5%~35%, 经还原后Cu粒子也仅为(3~8) nm。与尿素分解法不同, 如果向含有初始沉淀物Cu2(OH)3NO3和SiO2的体系中滴加KOH, 会导致碱式硝酸铜分解为CuO, 而非与SiO2反应生成强相互作用的铜化合物。
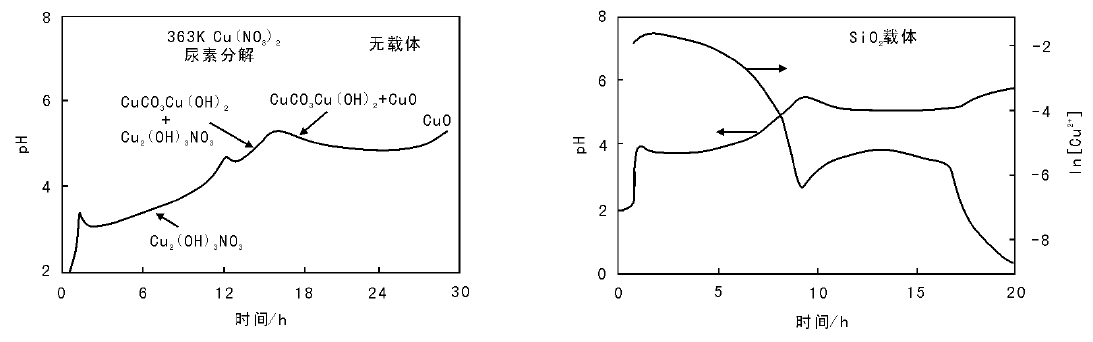 | 图 2-22 Cu(NO3)2沉淀过程的pH值变化[127] |
2.3.3.3 负载贵金属和金催化剂
负载贵金属催化剂多采用浸渍法制备, 但沉积-沉淀法也有其独特的优势, 易得到高金属负载量的催化剂且具有强金属-载体相互作用[131, 132]。通常沉积-沉淀操作可采取以下三种方式:(1) 向悬浮载体的贵金属盐溶液中滴加碱液; (2) 贵金属盐和尿素溶液中加入载体后通过尿素分解调控pH值[137]; (3) 向悬浮载体的碱性溶液中滴加贵金属溶液[134], 但此法应用较少。总的来说, 目前对大多数贵金属体系沉积-沉淀过程的机理缺乏深入的认识, 相对研究较多的是金催化剂的制备过程[135]。
通过滴加碱液使贵金属在载体表面沉积是应用最广泛的一种方法。通过向IrCl4溶液中滴加NaOH调节pH值到8(负载质量分数1.48%)制备的Ir/TiO2催化剂, 经673 K焙烧后发现, IrO2在TiO2表面形成厚度为2 nm的均匀薄层, 经氢气还原后转化为相同结构的Ir/TiO2催化剂; 当调节pH值分别为3.0(负载质量分数0.57%)和5.0(负载质量分数0.64%)时, 可得到粒径约为(2~3) nm的IrO2粒子和厚薄不均一的IrO2层。而采用浸渍法制备的催化剂IrO2粒子大小不均一, 大的IrO2粒子粒径达50 nm, 小粒子粒径5 nm[136]。将Na2CO3滴加到PdCl2和ZrO2的悬浮溶液中, 沉积-沉淀法制备出的Pd/ZrO2催化剂, Pd粒子的粒径约为1 nm, 与浸渍法制备的催化剂相比, 沉积-沉淀法制备的催化剂Pd与ZrO2间的相互作用更强, Pd带部分正电荷, 在氯苯加氢脱氯反应中, 该催化剂可以有效防止HCl吸附引起的催化剂中毒, 显著提高催化剂稳定性[137]。采用尿素分解调控溶液pH值, 通过逐渐升高pH值使金属盐沉积, 可以制备出高分散度的贵金属催化剂。以Pt(NH3)4(NO3)2为前驱体, 分别采用沉积-沉淀法和离子吸附法(ion adsorption)制备Pt/CNF(碳纳米纤维)催化剂。发现两种方法都可以制备出Pt平均粒径为(1~2) nm的高分散且热稳定性好的催化剂, 但当Pt投料比按质量分数5%计量时, 吸附法制备的催化剂Pt负载质量分数仅有2%, 而沉积-沉淀法可以高达4%。同样采用沉积-沉淀法以Ru(NO)(NO3)3为前驱体制备的Ru/CNF催化剂的Ru粒子平均粒径也仅为(1~2) nm(负载质量分数5%), Ru分散度高达74%。所制备的Pt/CNF和Ru/CNF催化剂经过773 K、N2处理后粒径仍然保持不变, 呈现优异的热稳定性[133]。可见, 沉积-沉淀法在负载贵金属催化剂制备中是浸渍法的有益补充, 某些情况下甚至可以制备出结构性能更优异的催化剂。
在纳米胶体金制备技术开发之前, 沉积-沉淀法曾是制备纳米金催化剂(< 5 nm)的典型方法之一(另一种方法为共沉淀法)。采用该方法制备的金粒子大小为(2~3) nm, 在CO氧化反应中表现出了优异的性能[139]。采用沉积-沉淀法制备金催化剂一般是通过向氯金酸(HAuCl4)和悬浮载体的溶液中加入碱, 控制溶液pH值为6~10, 溶液温度在(50~90) ℃搅拌下保持约1 h, 充分洗涤以除掉残留的Na+和Cl-(Cl-的存在会引起Au粒子的聚集)。常用的碱是Na2CO3和NaOH, 采用Na2CO3可以更好地控制溶液pH值。但该方法不大适用于等电点小于5的载体, 金难以在载体表面沉积, 因而形成较大的金粒子 [140]。
Au/TiO2催化剂的制备过程中, pH值对金的粒径有决定性作用。如图2-23所示, 以P25为载体, HAuCl4为金属盐前驱体, NaOH滴加控制溶液pH值; 随pH值升高, 金粒子尺寸由> 10 nm(pH< 5)迅速减小到< 5 nm(pH> 6)和3 nm (pH=7~10)。另一方面, 金的负载量随pH值变化呈火山型态势, 当溶液中加入计量比对应质量分数13%的HAuCl4时, 金的最高负载质量分数达到约8%(pH=5~6); 继续升高pH值到7~8时, 金的负载质量分数降低到3%左右。从而可以看出, 金不能全部从溶液中沉积到TiO2载体上。这可能因为HAuCl4在溶液中的存在状态与pH值密切相关, 可能的状态有AuCl3· H2O(pH=3~4)、AuCl2(OH
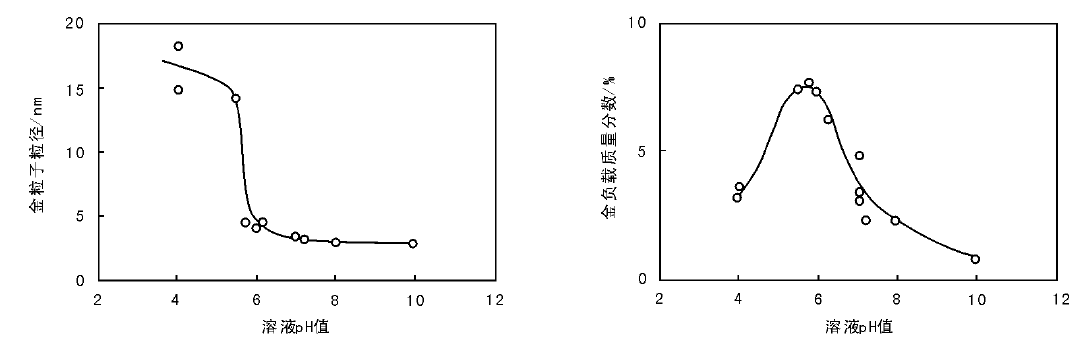 | 图 2-23 金粒子尺寸和负载量随pH值的变化 |
有报道认为Au在TiO2表面沉积-沉淀过程的机制与其他金属不同, pH=7~8时, Au在TiO2表面的存在形式并非Au(OH)3, 可能是通过如与TiO2表面反应形成表面络合物:TiOH+[AuCl(OH)3]-→ Ti-[O-Au(OH)3]-+H++Cl-。 这与X射线近边吸收谱(XAS)得到的数据吻合, TiO2表面沉积的Au物种周围没有Cl且近邻的氧有4个而非3个[143]。
早期研究结果表明, 尿素分解法控制pH值沉积-沉淀制备Au/TiO2催化剂得到的金粒子粒径较大(7.5 nm), 不如滴加NaOH或NaCO3溶液有效[144]。后来研究发现, 采用尿素分解法通过延长沉积-沉淀时间同样可以制备出金粒子平均粒径约为2 nm的催化剂, 且可以将Au从溶液近乎完全沉积在TiO2载体上。如表2-3所示, 采用尿素分解控制HAuCl4沉积-沉淀制备Au/TiO2催化剂过程中发现, 在初始的1 h内金已基本完全沉积在载体上, 此时溶液的pH值约为3.0, 在2 h时溶液的pH值升高到> 6, 此后并逐渐升高至7~8。在此过程中, 金粒子平均粒径由1 h的5.6 nm减小到4 h的2.7 nm, 此后金粒子粒径变化不大。此外, 整个过程中载体上沉积的金负载量仅在小范围内波动。尿素分解法中, 金物种沉积过程的机制与NaOH滴加法不同, 金在pH值为3左右即可完成沉积过程, 沉积物种的化学组成可能是AuN2.2O1.2C0.9H4.2Cl0.1, 它是由金与尿素分解的中间物形成, 初次沉积过程完成后, 表面金物种在体系内可能通过溶解再结晶过程实现二次分散[145]。
| 表 2-3 尿素分解法控制HAuCl4沉积-沉淀制备Au/TiO2催化剂[145] |
2.4 固体催化剂制备技术进展
2.4.1 微波和等离子体技术
微波加热:微波是频率介于300 MHz~300 GHz(波长1 m~1 mm)的超高频振荡电磁波, 在微波能量场作用下, 分子偶极矩与高频电磁波作用, 吸收微波能量变成热能。微波直接作用于分子, 使微波场中的整个介质同时被加热均匀, 因此微波加热是无温度梯度的“ 体加热” 或者说是内部加热[146]。与传统加热方式相比, 微波加热更均匀、更迅速, 可能制备出具有特殊结构和性质的催化材料。
微波加热在水/溶剂热合成催化材料中广泛应用, 该方式可加快晶化速率, 缩短合成时间。如采用微波加热方式合成Y分子筛时, 微波辐射约10 min即可完成晶化过程[传统加热方式(10~50) h], 且能有效避免P型分子筛等杂晶的生成[147]。一般认为, 这是由于微波对硅铝凝胶的分解有促进作用, 使凝胶快速溶解并形成大量晶核, 诱导晶体的生长[148, 149]。Cu/ZnO/Al2O3催化剂制备中, 在沉淀和老化阶段使用微波加热提高了催化剂活性和稳定性, 这可能是由于微波辐射在老化阶段促进了Cu2+对Zn5(CO3)2(OH)6中Zn2+的取代, 增加了沉淀物中(ZnCu)5(CO3)2(OH)6的含量[150]。负载型催化剂制备过程中, 也发现微波对活性组分的分散有促进作用。微波能显著促进ZnCl2与Y分子筛的固态离子交换过程[151], 微波辐射15 min即可使ZnCl2完全分散, 同时Na型分子筛比H型分子筛更易于发生固态交换, 这可能是由于Na+的存在能够促进分子筛对微波的吸收[152]; 乙二醇溶液中还原沉积-沉淀法制备CeO2、TiO2、ZnO和SiO2负载Pt催化剂过程中发现, Pt可以选择性沉积在CeO2、TiO2和ZnO表面[Pt粒径(2~3) nm], 而不能沉积在SiO2上, 这是由于CeO2、TiO2和ZnO对微波有较强的吸收, 使得固液界面处的温度高于溶液的温度, 促进了Pt的还原和选择性沉积, 而SiO2对微波吸收较弱[153]。微波加热应用于催化剂干燥过程发现, 微波辐射可以实现快速干燥, 有利于活性组分在载体上的均匀分布[154]。微波干燥法制备的Co/SiO2催化剂比常规干燥的催化剂有更高的F-T反应活性, 这是由于微波干燥促进活性组分在整个载体颗粒上的均匀分布, 活性组分分散度更高、粒子尺寸更加均匀, 而常规干燥法导致了Co在催化剂外表面的富集[155]; Ni基整体式催化剂制备过程中, 也发现微波干燥有效避免了活性组分在催化剂外表面的富集(图2-24)[156], 这可能是由于微波干燥有利于实现溶剂在整个载体颗粒上的均匀挥发[157]。微波加热还应用于催化剂的热处理/焙烧过程, 浸渍法制备的Pd-Fe/Al2O3催化剂(干燥后)采用微波辐射处理后发现, 与普通焙烧方式相比, 微波辐射导致金属粒子晶粒尺寸长大, 抑制了钯合金和低温下β -PdHx的生成, 催化剂在氯苯加氢脱氯反应中活性高于常规焙烧方法制得的催化剂[158]。
 | 图 2-24 常规干燥和微波干燥后活性组分在整体式催化剂上的分布(截面图)[156] (a)实物图, (b)示意图 |
等离子体技术:等离子体被称为物质的第四态, 是由气体分子在受热或外加电场及辐射等能量激发下解离、电离形成的离子、电子、原子、分子和自由基等的集合体。等离子体整体呈电中性, 是物质的一种高能存在状态。等离子体可分为低温等离子体和高温等离子体, 低温等离子体技术在催化剂制备中应用较多[159, 160]。
在负载型催化剂制备中, 等离子常用于负载型催化剂活性组分前驱体的分解过程[161]。在Ir/Al2O3催化剂制备过程中, 采用等离子体处理浸渍H2IrCl6的催化剂, 可将Ir物种还原为金属态, 所得催化剂在Ar气氛中经600 ℃处理3 h后, Ir粒子仍可保持在1 nm, 而传统氢气还原得到的催化剂Ir粒子为3 nm[162]。等离子体溅射技术可用于金属/氧化物的化学气相沉积过程, 制备负载金属/氧化物催化剂。以金属为阳极采用脉冲电弧-等离子体蒸发可以将金属沉积在载体表面, 制备出分散度高且粒径均一的催化剂, 如以等离子蒸发制备Rh/AlPO4催化剂, Rh粒子约为(2.4± 1.1) nm, 而以浸渍法制备的催化剂Rh粒子大小为(6.4± 5.5) nm[163]。采用双/多脉冲法还可以制备双/多金属催化剂, 通过脉冲调谐可以控制各金属组分的含量以及金属间的作用形式, 如同步脉冲法可以得到单个金属粒子同时含有钯和铁的Pd-Fe/CeO2催化剂, 而顺次脉冲法得到的是钯粒子和铁粒子独立存在的催化剂, 两个催化剂上金属粒子大小均为(2.3± 0.7) nm。在单个金属粒子同时含有钯和铁的催化剂(同步脉冲法)上, 金属态钯和铁的含量均高于钯和铁粒子独立存在的催化剂体系(顺次脉冲法)[164], 表明钯和铁的结合改变了其存在的化学状态。
等离子还常用于催化剂结构的修饰和改善。采用低温等离子体在Ar、H2和NH3气氛中对锐钛矿相TiO2纳米片(主要暴露{001}晶面)进行了处理, 发现催化剂表面氧缺陷位(或Ti3+)增多, 形成了TiO2@TiO2-x核壳结构, 增强了催化剂的近红外光吸收[165]。采用辉光放电等离子体对天然斜发沸石进行处理, 样品形貌由不规则粒子变成直径< 100 nm的棒状结构(图2-25), 比表面积由24 m2· g-1增加到45 m2· g-1, 并产生介孔, 催化剂孔结构进一步开放[166]。此外, 使用等离子体轰击金属盐、氢氧化物、金属等, 使之分解或与气体(如氧气)反应可以制备纳米粉体, 如采用等离子体轰击Fe、Al、K2CO3、CaCO3和SiO2混合粉末可制备合成氨催化剂[167]; 在30%O2-Ar气氛中轰击Ni、Al粉末可制备Ni-O-Al天然气重整催化剂[168]。
 | 图 2-25 天然斜发沸石和等离子体处理后样品的SEM照片[166] |
2.4.2 原子层沉积法
原子层沉积(ALD)也称为原子层外延生长(ALE), 其原理是利用气相分子与固体表面官能团间的自消除反应(self-limiting reaction)在固体表面形成均匀单原子层结构化合物[157, 169, 170, 171]。一般原子层沉积采用易挥发的金属有机物作为沉积前驱体分子, 所谓自消除反应是指前驱体分子与载体表面官能团(一般为羟基)发生类似缩合反应的过程。如钛酸四异丙酯[Ti(OiPr)4]沉积在SiO2表面时, 异丙氧于SiO2表面羟基反应脱除异丙醇。
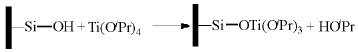
随Ti物种在SiO2表面覆盖度增加, SiO2表面羟基被消耗, 而Ti-OiPr对Ti(OiPr)4是反应惰性的, 因此, 原子层沉积的自消除反应特性使得金属(气相中的前驱体分子)在载体表面上的单原子层沉积成为可能。而从广义上讲, 原子层沉积方法还包括后续的系列反应过程, 初次沉积完成后, 向反应体系内通入其他气体(如氧气、水蒸气、氨气等)与沉积物继续反应, 这一过程在固体表面重新形成具有反应活性的官能团(如上述SiO2表面沉积的钛物种与H2O反应), 利用这些官能团可以进一步通过自消除反应再次进行原子层沉积过程, 由此可以实现金属(或不同金属)在载体表面逐层沉积。
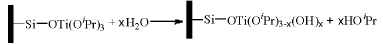
ALD广泛应用于负载型金属/金属氧化物催化剂制备。采用ALD技术制备负载型催化剂能够精确控制活性组分的组成、活性位密度以及粒子大小等, 且易于实现活性组分在催化剂表面和孔道内的均匀分布。活性组分在载体上的沉积状态受金属前驱体分子结构性质、载体孔道结构、载体表面活性反应官能团反应活性和分布状态(密度、均匀程度)[172]以及沉积反应温度等因素的影响[173]。气相中分子首先要扩散到载体表面和孔道内, 分子大小、蒸气压和载体的孔道结构会影响分子的扩散行为。如将Al(CH3)3和Zn(C2H5)2等易挥发的化合物沉积到多孔SiO2气溶胶孔道内, Al和Zn物种可以穿透到几百微米深度的SiO2整体式(monolithic)结构中, 形成均匀沉积; 而N, N’ -双-仲丁基乙酰基双铜{Cu(Ⅰ ) N, N’ -di-sec-butylacetamidinate, [Cu(sBu-Me-amd)]2}只能沉积到几十微米的深度。这是由于前驱物分子的扩散能力差异较大所致, Al(CH3)3和Zn(C2H5)2分子较小, 蒸气压可以达到约1.33 KPa, 而[Cu(sBu-Me-amd)]2的蒸气压不足13.3 Pa。此外, 前驱体分子的反应活性以及前驱体分子和产物分子在孔道内的吸附-脱附平衡也可能对沉积过程的穿透深度造成影响[174, 175]。载体表面, 特别是参与ALD过程自消除反应的活性官能团的反应活性, 是影响沉积反应速率的另一重要因素。在负载Pd催化剂制备过程中, 200 ℃下以六氟乙酰丙酮钯[Pd(Ⅱ ) hexafluoroacetylacetonate, Pd(hfac)2]和福尔马林蒸汽顺次流过载体表面进行沉积(循环多次), 结果发现, Pd在TiO2载体表面的沉积速率大于在Al2O3上的沉积, 而在SiO2上几乎不能发生沉积, 这可能是由于载体表面羟基的反应活性不同造成的[172]。同时发现, 如果沉积过程在110 ℃进行, 即使进行100次循环沉积, Al2O3表面仍然只有很少的Pd沉积。这可能是由于降低沉积温度后, Pd(hfac)2分解产生的hfac在载体表面吸附形成Al(hfac)* 物种[176], 毒化了载体表面。
ALD是一种投影式包覆(conformal coating)技术, 即能在固体表面形成与基底具有相同拓谱结构的包覆, 不会改变载体比表面积、孔道结构等[170], 因此ALD也常用于载体和催化剂表面的修饰。在SiO2和Al2O3等具有大比表面积的常用载体表面, 采用ALD沉积TiO2、ZrO2、CeO2等, 可以使新载体在基本保留基底孔道结构等物理性质的同时兼具沉积物的化学性质, 为催化剂设计和性能调控提供了便利。利用ALD技术在Al2O3表面沉积TiO2和CeO2后再沉积Pt, 发现与未沉积TiO2和CeO2修饰的催化剂相比, Pt粒子的分散度明显提高, Pt粒子大小由(3.4± 2.6) nm减小到(2.1± 0.5) nm(TiO2)和(1.6± 0.4) nm(CeO2)[177]。最近, 采用ALD技术对负载金属催化剂进行修饰, 通过在金属纳米粒子外包裹一层氧化物, 抑制金属在高温处理和催化反应过程中的聚集, 有效提高了催化剂稳定性[178, 179, 180, 181]。将Cu/Al2O3催化剂顺次暴露在三甲基铝和水蒸汽气氛(130 ℃, 循环45次), 可在铜粒子表面包覆一层铝氧化物。包覆后Cu的比表面积由86 μ mol· g-1降到0, 说明ALD技术实现了对Cu粒子的完全包覆。催化剂经973 K高温焙烧后, Cu的比表面积变为23 μ mol· g-1, 这是由于焙烧后包覆层产生了孔结构, 探针分子(N2O用于表征Cu的表面积)可以通过这些孔道接触到Cu表面。催化剂用于2-呋喃甲醛的液相加氢还原反应(130 ℃, 2.2 MPa)后, Cu粒子大小由(3.0± 1.0) nm长大到(5.0± 2.0) nm, 而经过包覆处理的催化剂Cu粒径基本保持不变[181]。利用类似的方法, 还可调控出Pd/Al2O3催化剂[Pd粒子尺寸(2.8± 0.5) nm]的高温稳定性。当Al2O3在Pd粒子表面包覆厚度达到8 nm时, 所得的催化剂在乙烷氧化脱氢反应(675 ℃)中表现了更好的稳定性, 近30 h反应的活性不变; 未经氧化铝包覆的催化剂初始活性只能保持10 min左右。这种稳定性的提高主要归因于Al2O3包覆提高了Pd粒子抗烧结能力和改善了催化剂的抗积炭能力。未经氧化铝包覆的催化剂的Pd粒子在反应30 min后长大到(4.6± 1.9) nm(粒径分布变宽), 同时催化剂表面生成纳米碳纤维将Pd粒子从载体表面剥离; 而包覆后的催化剂Pd粒径基本保持不变(图2-26), 积炭量大幅度降低。这可能是由于铝物种沉积时优先覆盖了Pd粒子表面低配位的Pd原子位置, 而这些低配位的Pd原子是C— C键断裂的活性中心, 造成CH4、CO和CO2等物种的生成(对照试验发现, 甲烷的存在是生成积炭的重要原因, 乙烷和丙烷等不会导致积炭生成)[178]。
 | 图 2-26 催化剂的STEM照片 (A)新鲜催化剂和(B、C)30 min反应后; (D)Al2O3包覆的Pd/Al2O3反应前和(E)1 700 min反应后; (F)Pd粒径分布 |
2.4.3 微/纳乳液法
2.4.3.1 乳液的形成
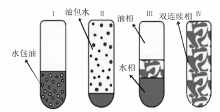
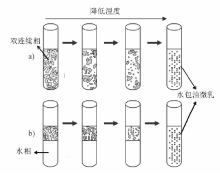
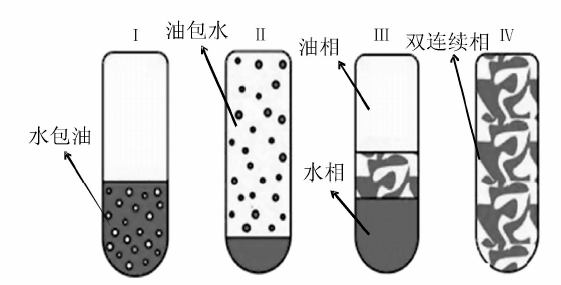
微乳液是由互不相溶的两种液体与表面活性剂及助表面活性剂在适当比例下自发形成的透明或半透明、各向同性热力学稳定均匀分散体系。互不相溶的两种液体分别称为水相(极性液体)和油相(非极性液体), 表面活性剂也称为乳化剂, 多为非离子型表面活性剂, 而助溶剂多为醇类。近年来, 有人提出纳米乳液(纳乳)的概念, 建议将纳乳液滴的大小定义到500 nm以下[182]或更小(< 200 nm)[183, 184], 并提出纳乳是热力学不稳定体系[185], 以此与微乳液的概念进行区分, 但作为一种材料合成方法多数文献对纳乳和微乳并不严格区分[186, 187], 一般仍称为微乳法。宏观上看微乳液是均匀的, 但它实际上是一种多相分散体系, 将油相分散在水相中称为水包油型微乳, 反之为油包水型微乳, 后者通常也称为反相微乳; 还有一类微乳体系称为双连续相微乳。
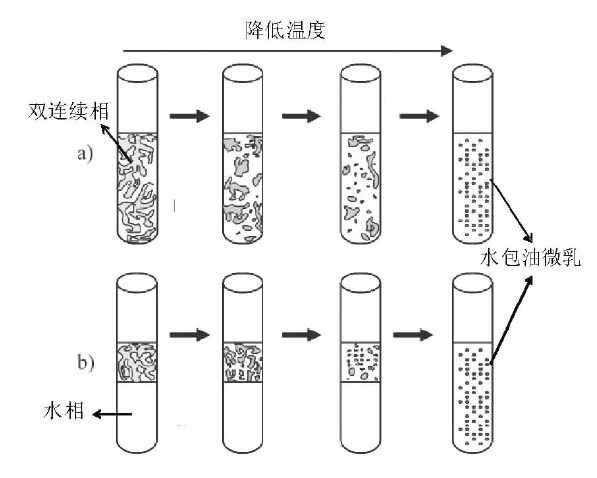
油、水和表面活性剂混合后, 体系存在状态(平衡态)分为四类, 即通常所说的四类Winsor体系(图2-27)[188]:(1) 油相过量, 油相+水包油型乳液(Winsor Ⅰ ); (2) 水相过量, 水相+油包水型乳液(Winsor Ⅱ ); (3) 油相和双连续相乳液或水相和双连续相乳液或三相共存(Winsor Ⅲ ); (4) 双连续相乳液(Winsor Ⅳ )。形成微乳液的方式有低能量乳化和高能量乳化两种。低能量乳化是指在不需要向体系输入大量的额外能量的情况下使乳化过程自发进行, 比较经典的是相反转温度法(phase inversion temperature-PIT method)。该法是利用表面活性剂在亲水亲油平衡温度(hydrophilic lipohilic balance-HLB temperature)时极低的界面能, 通过变化体系温度促进乳液的形成 [183, 188]。多用于聚氧乙烯(polyoxyethlene, PEO)型表面活性剂的体系[182], 低温时PEO亲水性强, 体系存在状态为Winsor Ⅰ 型; 随温度升高PEO亲油性增强, 达到HLB温度时体系存在状态为Winsor Ⅲ 或Winsor Ⅳ (与各组分含量有关); 高温时PEO亲油性强, 体系存在状态为Winsor Ⅱ 。在体系处于HLB温度时, 各组分含量合适的情况下, 通过快速升温或降温可以相应得到油包水和水包油型乳液。如Winsor Ⅳ 型和水+双连续相型乳液体系通过快速降温, 可以形成水包油型微乳液(图2-28)。同样道理, Winsor Ⅳ 型和油+双连续相型体系通过快速升温可以形成油包水型微乳液。恒温条件下, 通过改变物料配比(也称为phase inversion composition-PIC)也可以实现乳化, 常用的方法有溶剂置换法(solvent displacement method)和乳液反转点法(emulsion inversion point-EIP method)等。溶剂置换法利用有机溶剂(如丙酮、乙醇)由油相向水相定向快速扩散(Ozuo效应)形成乳液[189], 如向1∶ 100的二乙烯基苯/乙醇溶液中倾入水, 当乙醇质量含量在20%~40%时可以自发形成稳定的水包油乳液[190]。乳液反转点法是通过向热力学稳定的双连续相乳液体系内加入油或水进行稀释, 将其转变成液滴更小的动力学稳定乳液[182]。
 | 图 2-27 四类Winsor体系示意图[188] |
 | 图 2-28 PIT乳化法示意图[183] |
高能量乳化法需要通过剧烈的搅拌、超声或高压等方式向体系提供能量来制造界面区域引发乳化。以超声方式为例, 在超声作用下, 油相或水相被分散成很小的液滴, 然后表面活性剂在液滴界面上吸附使液滴稳定, 最终形成微乳液。输入的能量能否有效用于油相或水相的分散是决定乳化成功与否的关键, 如普通搅拌方式不如超声方式有效是由于前者输入到体系的能量大部分以热量的形式耗散掉, 没有起到将油相或水相变成小尺寸液滴的效果。
微乳液的两个重要特征是液滴大小(以及液滴粒径分布)和乳液的稳定性, 两者都与表面活性剂的本身化学性质、浓度、油/水比例、温度以及采取的乳化方式等有关系。显然, 低能量乳化法中, 乳液体系的组成比例和油、水、表面活性剂的性质是主要因素, 而高能量乳化方式还严重依赖于仪器(超声仪、高压匀质机等)对液滴的分散效率。总之, 乳液体系和乳液的形成机制非常复杂, 在微乳液的制备及其性质(液滴大小、乳液稳定性)的优化过程中, 实验手段仍发挥重要作用[185]。
2.4.3.2 微乳液法制备催化剂
固体催化剂的制备多以水溶性的无机盐为前驱体, 因此常用到反相微乳(油包水)体系。在反相微乳体系中, 水相在表面活性剂包覆下以纳米级[如(10~100) nm]“ 水核” 的形式分散在油相中, 作为“ 纳米反应器” 限定固体的成核和生长。“ 水核” 作为纳米反应器合成固体的方式一般有两种, (1) 双乳液法:金属盐和沉淀剂(合成金属纳米粒子时使用还原剂)分别配成两种微乳体系, 混合搅拌后包含两种反应物的微乳液滴间碰撞结合导致“ 水核” 间的物质交换而引发化学反应; (2) 单乳液法:将一种反应物溶解在微乳“ 水核” 中, 另一种反应物(直接或溶解后)加入到微乳液体系内, 然后通过微乳液界面渗透到“ 水核” 内或直接在界面处发生反应; 或将反应物全部溶解在“ 水核” 中, 通过改变反应温度使之反应[187, 191, 192]。通过控制反应在乳液液滴内部或界面处发生可以合成纳米粒子或空心结构, 调控液滴大小可以控制催化剂尺寸和形貌[193]。利用微乳液法合成金属或金属氧化物纳米粒子催化剂的研究很多, 以往的文献作了很好的总结[187, 194], 此处仅对微乳法在纳米空心结构和空心内含纳米粒子结构催化剂制备方面的近期工作做简单介绍。
 | 图 2-29 微乳液法制备的Au、AlO(OH)、和SnO2空心结构HRTEM照片[192] |
空心结构的合成需要控制反应在微乳液滴的界面处发生。通常将一种反应物溶解在微乳液滴内, 然后向主相内加入另一种反应物, 反应主要受扩散控制, 固体选择性的沉积在液滴界面处。为此, 需要针对反应物的溶解性、浓度、物料滴加速率、反应温度以及反应速率等进行实验筛选和条件优化[191]。目前利用微乳液法制备了Ag、Au、ZnO、γ -AlO(OH)、TiO2、SnO2等纳米空心结构。合成La(OH)3空心纳米结构的过程中, 以正十二烷(50 mL)、十六烷基三甲基溴化铵(CTAB, 5 mmol)和正己醇(5 mL)分别作为油相、表面活性剂和助表面活性剂, 通过控制水的加入量[(1~3) mL], 在剧烈搅拌下(10 min)成功获得了液滴大小(5~12) nm的微乳体系, 然后向体系内加入三环戊二烯基镧[La(Cp)3]的十二烷溶液, 与水核反应生成La(OH)3。水相为纯水时得到纳米粒子, 而在水相中加入一定浓度(0.1 mol· L-1或0.2 mol· L-1)卤化钾(如KF)则可得到空心结构。这是由于盐的加入使La(Cp)3的水解变慢, 降低了La(OH)3的过饱和度, 避免了高过饱和度下的均匀沉淀, 使沉淀过程选择性发生在界面处 [195]。利用同一乳液体系, 还制备了γ -AlO(OH)和Ag空心纳米结构; 前者选择Al(sec-OC4H9)3为铝源前驱体, 是由于其水解速率较慢且在油相中的溶解度大于水相中的溶解度; 后者以[Ag(PPh3)4]NO3为前驱体, 并将含有NaBH4的水核pH值调节为12, 以降低还原反应速率; 其目的都是要利用扩散控制的水解(diffusion-controlled hydrolysis)/还原反应使沉积过程发生在界面处[196, 197]。
在空心结构内植入纳米粒子还能得到空心内含有纳米粒子型的结构, 合成的策略主要有三种:(1) 先将制备好的纳米粒子分散在乳液液滴内, 再以液滴为模板合成空心结构; (2) 乳液液滴内预先溶解合成纳米粒子的前驱体, 空心结构形成后再将液滴内的前驱体转化成纳米粒子; (3) 先将微乳液滴内溶解的化合物转化成纳米粒子, 再以液滴为模板合成空心结构。纳米粒子的植入需要结合空心结构的合成, 选择合适的纳米粒子(或前驱体盐)和与之匹配的乳液体系, 并通过巧妙实验设计充分利用反应过程中的物理化学变化。如采用反相微乳法合成Fe3O4@SiO2过程中, 正硅酸乙酯(TEOS)加入微乳体系前Fe3O4在油胺保护下处于油相中, TEOS水解后置换掉Fe3O4表面的油胺将Fe3O4转移到水相, 即微乳液滴内, 然后TEOS的水解和沉积形成空心SiO2(壳层)内植入Fe3O4的结构[198]。在上述过程中, 如果“ 水核” 中溶解了易还原的金属盐, 如HAuCl4, 其可在形成空心结构的同时被表面活性剂还原并沉积在Fe3O4纳米粒表面。如果再加入NaBH4, Fe3O4可以在金辅助下还原为Fe2+而溶解掉, 最终形成Au@SiO2结构[199]。
2.5 结语和展望
固体催化剂的制备不仅仅是一个单纯的化学反应问题, 催化剂结构的调控更多的涉及到纳米材料的物理化学问题。本章仅集中对部分溶液化学合成方法, 从物理化学的视角对制备过程的变化机制进行了阐述, 确切的说主要涉及催化剂前驱体/前驱物(precursor)的制备, 而对催化剂的焙烧、活化、成型等后续过程基本没有涉及。近年来, 纳米材料领域的突破引发了新结构催化剂合成研究热潮, 新合成方法不断涌现, 对催化剂微观结构的控制更加精准, 手段更加丰富, 极大的推进了对催化剂结构和催化性能之间的构效关系的理解。但需要指出的是, 与材料合成和催化剂结构表征研究形成鲜明对比的是, 对材料合成的溶液物理化学和固液界面物理化学的系统研究较少, 动力学方面的理解和认识相对滞后, 对很多现象的解释仍然停留在原有的经验性理论和概念。因此, 对催化剂制备和材料合成过程微观物理和化学变化原理的认识尚有待深入。

申文杰, 中国科学院大连化学物理研究所研究员、博士研究生导师。从事催化材料和催化反应过程的应用基础研究。主要针对能源和环境相关的催化过程, 如生物质制氢、甲烷、甲醇/二甲醚转化、NOx和VOC脱除等, 开展催化材料制备化学、结构分析表征、反应机理及动力学等方面的工作。在催化材料设计和合成方面, 重点以调控催化活性组分的尺寸和形貌为导向, 开展催化剂制备的溶液化学、粒子尺寸和形貌的可控合成、金属-载体相互作用机制等方面的系统研究。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
| [169] |
|
| [170] |
|
| [171] |
|
| [172] |
|
| [173] |
|
| [174] |
|
| [175] |
|
| [176] |
|
| [177] |
|
| [178] |
|
| [179] |
|
| [180] |
|
| [181] |
|
| [182] |
|
| [183] |
|
| [184] |
|
| [185] |
|
| [186] |
|
| [187] |
|
| [188] |
|
| [189] |
|
| [190] |
|
| [191] |
|
| [192] |
|
| [193] |
|
| [194] |
|
| [195] |
|
| [196] |
|
| [197] |
|
| [198] |
|
| [199] |
|