{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
第四章 石油炼制催化作用
引用本文
蒋宗轩, 刘欣毅. 第四章 石油炼制催化作用[J]. 工业催化, 2016,24(1): 84-112. 
Permissions
Copyright©2016, 《工业催化》编辑部
《工业催化》编辑部 所有
第四章 石油炼制催化作用
4.1 前 言
如今, 石油炼制工业已成为我国经济的重要支柱产业, 在能源和有机化工等领域占有重要地位。据统计, 全世界约有40%的能源需求依赖于石油产品, 而有机化工原料也主要来自石油产品, 世界每年生产的石油约有10%用于有机化工。由于石油是多种有机化合物的混合物, 组成复杂, 其相对分子量分布从几十至几千, 对应的沸点也从常温到500 ℃以上, 不能直接使用, 必须经过复杂的加工炼制, 才能成为不同用途的各种石油产品。目前已有的石油产品种类繁多, 包括各种燃料油(如柴油、汽油)、润滑油、有机化工原料、沥青、蜡及石油焦等, 可见人们的生活已经离不开各种各样的石油产品。
石油炼制工业最早是通过简单的蒸馏用于生产家用煤油。到20世纪初, 由于第一次世界大战和汽车工业的发展, 导致汽油需求量猛增, 通过简单蒸馏得到的汽油产量已经不能满足人们的需求, 因此使用重馏分油生产汽油的裂化技术由此诞生, 并且在20世纪40年代逐渐成为生产汽油的主要方法。到了50年代, 为了满足对汽油抗爆性的要求, 又出现了铂重整技术, 大大促进了重整技术的发展。同时由于重整技术带来了廉价的氢气副产品, 又带动了加氢技术的发展。到了60年代, 由于分子筛催化剂的出现和发展, 导致催化裂化技术发生了革命性的变革。70年代, 由于石油来源和价格等因素, 又促进了重质油轻质化技术的发展。相对于世界其他发达国家, 我国的石油炼制工业发展较晚, 1958年才建立了新中国第一座炼油厂。到60年代, 在大庆油田的发现和开发带动下, 我国石油炼制工业开始迅速发展。目前我国炼油工业居世界第二位, 炼油技术水平也进入世界先进行列[1]。
目前, 原油重质化已经成为世界炼油工业面对的一大问题。由于我国原油多数偏重, 多数原油中减压渣油含量在40%以上, 因此这一问题对于我国炼油工业更具有特殊性和重要性。为了解决该问题, 重质油轻质化技术受到越来越多的关注。另一方面, 随着各国对环境保护要求的逐渐提高和对燃料油污染物排放限定要求的严格化, 也对我国炼油技术提出了新的挑战。
本章将对石油的组成和性质作简要介绍, 并结合我国国情和炼油工业的发展历史, 从最具代表性的催化裂化、加氢裂化、加氢精制和催化重整等方面, 展示石油炼制工业中涉及到的催化科学。
4.2 石油的性质及化学组成
石油通常是黑褐色的黏稠液体, 相对密度为0.80~0.98。不同产地的原油性质有所差异, 与外国的原油相比, 我国的原油相对密度偏大, 凝点偏高, 属于偏重的原油。
石油是组成复杂的混合物。虽然不同产地的石油组成有所不同, 但所含元素大致相同, 碳、氢、氧、硫、氮占据其组成的99%以上, 其中95%来自碳和氢组成的各种脂肪族和芳香族烃类化合物。由于不同类型的有机烃类化合物氢碳原子比不同, 因此石油的氢碳原子比可以反映出石油的属性。在石油加工过程中, 通过催化手段使有机分子发生键断裂和氢转移反应(裂化反应和重整反应), 可以得到不同的氢碳比较高的轻质产物和氢碳比较低的重质产物。除了碳、氢之外, 石油中还含有氧、氮、硫和一些其他微量元素, 这些元素虽然含量很少, 却对石油的性质和加工过程有很大影响。同外国原油相比, 含硫低、含氮高是我国原油的特点之一。
对于组成复杂的石油, 其沸点范围很宽, 要想对石油进行加工利用, 首先必须按照沸点的不同将石油分割成不同的馏分, 这个过程也称作石油的分馏。一般将石油在常压下从初馏点到200 ℃的馏分称为汽油馏分(或石脑油馏分), (200~350) ℃的馏分称为煤柴油馏分(或常压瓦斯油、AGO), (350~500) ℃的馏分称为润滑油馏分(或减压瓦斯油、VGO), 高于500 ℃的馏分称为减压渣油(VR)。其中, 需要注意的是为了保证石油在分馏时不发生分解反应, 对于沸点高于350 ℃的馏分, 必须在减压条件下进行蒸馏, 然后再将减压条件下得到馏分的沸点换算成常压下的沸点。同国外石油相比, 我国原油中的减压渣油含量较高, 而汽油馏分含量较少。从石油直接分馏得到的馏分称为直馏馏分, 其性质与原来的石油相似; 而经过二次加工所得到的馏分在组成上则与直馏馏分有所区别[2]。
4.3 催化裂化及加氢裂化中的催化作用
石油经过常压、减压蒸馏可以得到汽油、煤油和柴油等轻质油品, 但收率和质量并不高。随着工业的发展, 对轻质油品的数量和质量提出了更高的要求, 而催化裂化和加氢裂化是获得更多轻质油品并提高其质量的重要二次加工过程。
4.3.1 催化裂化反应
催化裂化是重质石油烃类在催化剂存在时, 于(470~530) ℃和(0.1~0.3) MPa条件下, 发生以裂解反应为主的一系列化学反应, 转化成气体、汽油和柴油、重质油及焦炭的工艺过程。催化裂解的主要目的是将重质油品转化成高质量的汽油和柴油等产品, 是重质油轻质化的主要手段。
催化裂化原料范围很广, 有(300~500) ℃直馏馏分油、常压渣油和减压渣油, 也有二次加工馏分。我国原油直馏馏分油具有以下特点:(1) 原油中轻组分少, 催化裂化原料充足; (2)含硫低, 含重金属少。因此, 直馏馏分油是理想的催化裂化原料。对于二次加工馏分, 具有芳烃含量大, 裂化性能差, 一般不能单独作为催化裂化的原料。而对于常压渣油和减压渣油, 由于我国原油大部分为重质原油, 减压渣油收率约占原油的40%, 常压渣油约占70%, 因此重油裂化工艺可以提高原油加工深度, 有效利用石油资源[3, 4]。
通常用以下几个指标来评价催化裂化原料的性能:
(1) 馏分组成:可以判别原料的轻重和沸点范围的宽窄, 馏分越重越容易裂化;
(2) 烃类组成:用来表示烷烃、环烷烃和芳烃的含量, 但重质原料油烃类组成分析困难, 一般用密度、特性因数K(K值高说明含烷烃多, K值低说明含芳烃多)和苯胺点(苯胺点越低则芳烃含量越高)来进行间接判断;
(3) 残炭:原料残炭值越高则生焦越多;
(4) 金属:原料油中重金属对催化剂的活性和选择性有很大影响, 在催化裂化过程中沉积在催化剂上, 引起催化剂中毒失活;
(5) 硫、氮含量:原料中的含氮化合物含量多时会引起催化剂中毒, 此外还会造成产品油变色和氧化安定性变坏。而含硫化合物会增加设备腐蚀, 使产品硫含量增高, 污染环境。因此如果催化裂化使用的原料中硫和氮含量较高则应当进行预精制处理。
催化裂化过程中使用的原料、催化剂和反应条件不同, 所得产品的产率和性质也不同。一般工业条件下, 气体产率约为10%~20%, 其中主要为C3和C4烃类组分, 还有少量的C1、C2组分和氢气、硫化氢。液体产品中催化裂化汽油产率为40%~60%, 由于其中含有较多的烯烃、异构烷烃和芳烃, 所以辛烷值较高, 约为80; 并且由于基本不含二烯烃, 安定性也比较好。而柴油产率为20%~40%, 由于含有较多的芳烃, 所以十六烷值比直馏柴油低, 通常与直馏柴油等调和后才能作为燃料油使用。此外还有少量焦炭沉积在催化剂上, 其产率随原料质量不同而异。
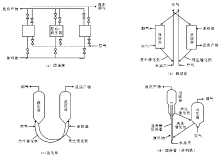
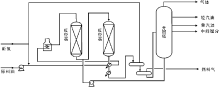
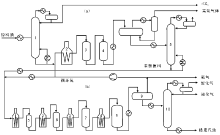
催化裂化技术方法主要分为固定床、移动床、流化床和提升管反应器。固定床的反应和再生过程在同一设备中交替进行, 属于间歇式操作(图4-1a), 这种装置的设备结构复杂, 生产能力低, 已被淘汰。移动床催化裂化使用直径约3 mm的小球催化剂, 由空气提升, 其生产能力比固定床大有提高(图4-1b), 但其设备结构比较复杂, 钢材耗量也比较大。流化床催化裂化在最近几年得到迅速发展, 使用(20~100) μ m的微球催化剂, 在反应器和再生器内与油气或空气形成流化状态, 在反应器和再生器之间循环, 具有处理量大, 设备结构简单等优点(图4-1c), 但由于存在床层返混现象, 产品质量和产率不如移动床。随着20世纪中期分子筛催化剂的问世, 为了使分子筛催化剂的高活性得以发挥, 流化床反应器逐渐被提升管反应器取代(图4-1d)。提升管反应器以高温段接触时间的活塞流反应代替了原来的床层反应, 因而克服了床层返混, 使得产品质量和产率得到大幅提高。
催化裂化装置主要由反应-再生系统、分馏系统等组成, 其中反应-再生系统是催化裂化装置的核心, 任务是使原料油通过反应器或提升管与催化剂接触反应生成产物, 并且不断地将催化剂送入再生器中烧去焦炭再生后送回反应器。分馏系统主要由分馏塔、柴油汽提塔, 原料油缓冲罐塔顶油气冷凝冷却回流系统等组成, 其主要任务是将来自反应系统的高温油气脱热过后根据沸点的不同切割为不同的馏分。
1936年世界上第一套固定床催化裂化工业化装置问世, 成为了催化裂化工艺的开端。之后在40年代相继出现了移动床催化裂化装置和流化床催化裂化装置, 60年代又出现了提升管反应器。70年代之后, 分子筛催化剂又继续向着高活性、高耐磨、高环保性的方向发展。通过多年来的生产实践和技术探索, 我国已经掌握了整套重油催化裂化技术和多种催化裂化工艺, 并且还在不断发展中。这些工艺不仅推动了催化裂化技术的进步, 而且也满足了炼油厂对新产品结构和质量的要求[5, 6]。
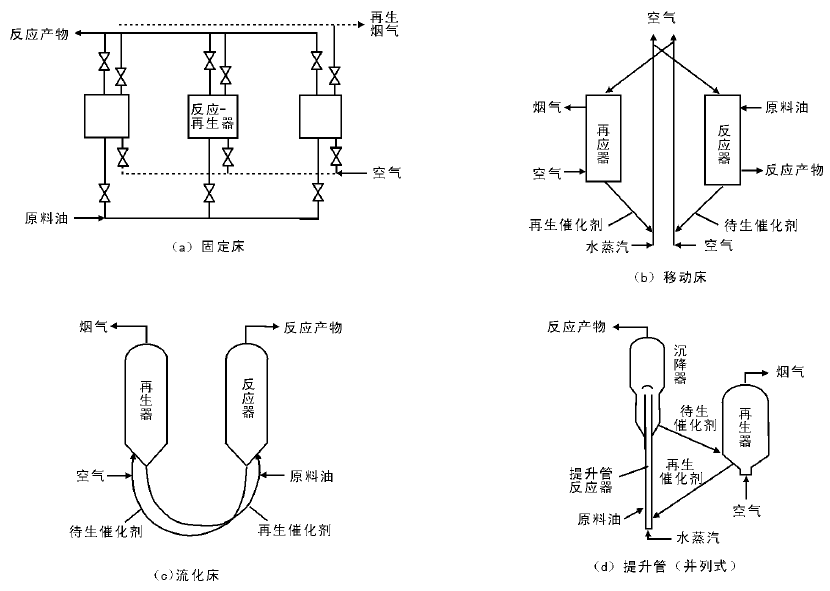 | 图 4-1 催化裂化反应-再生系统的几种型式 |
催化裂化产品的数量和质量, 取决于原料中的各类烃在催化剂上的反应。要想更好的控制生产, 就必须先了解催化裂化反应的本质、特点以及影响因素。石油馏分中的各种烷烃、环烷烃和芳烃在催化剂上进行不同的反应, 以分解反应为主, 还有异构化反应、氢转移反应、芳构化反应等。为了更好地了解催化裂化的反应过程, 首先了解单体烃的催化裂化反应。
(1) 烷烃:烷烃主要发生分解反应, 生成较小分子的烷烃和烯烃:
C16H34→ C8H16+C8H18
生成的烷烃又可以继续分解。烷烃分解时一般从中间的C— C键处断裂, 而且分子越大越容易断裂。对于碳数相同的链状烃, 异构烷烃的分解速率比正构烷烃快。
(2) 烯烃:烯烃的主要反应也是分解反应, 分解为两个较小分子的烯烃, 其速率比烷烃的分解速率快得多:
C16H32→ C8H16+C8H16
烯烃还会发生异构化反应, 如双键移位异构和骨架异构:
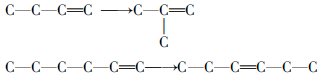
另外烯烃还会发生氢转移反应和芳构化反应, 其中氢转移反应是指烯烃将氢转移给其他烯烃分子使之饱和, 而芳构化反应是指烯烃环化脱氢生成芳烃:
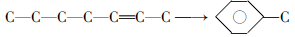
(3) 环烷烃:环烷烃可断裂生成烯烃, 也可脱氢成为芳烃:
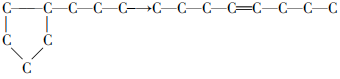
(4) 芳烃:芳烃在催化裂化条件下十分稳定, 一般主要是烷基侧链容易发生裂解反应。多环芳烃裂化速率很慢, 一般缩合为稠环芳烃再转化为焦炭。
催化裂化反应是非均相反应, 原料进入反应器气化后, 首先向催化剂表面扩散, 再在催化剂表面吸附, 然后再进行化学反应。对于碳原子数相同的各类烃吸附顺序为:稠环芳烃> 稠环环烷烃> 烯烃> 单环芳烃> 环烷烃> 烷烃。而化学反应速率的顺序则为:烯烃> 大分子单环芳烃> 异构烷烃、环烷烃> 小分子单环芳烃> 正构烷烃> 稠环芳烃[7, 8, 9]。
4.3.2 催化裂化催化剂
4.3.2.1 催化裂化催化剂的组成与结构
工业上使用的催化裂化催化剂主要可以分为三类:天然白土催化剂、无定形合成催化剂和分子筛催化剂, 其中现在广泛使用的是分子筛催化剂。
工业催化裂化装置最初使用的催化剂是经过处
理的天然活性白土, 其主要成分是硅酸铝。之后, 天然活性白土就被人工合成硅酸铝所取代。而这两种催化剂都是无定型硅酸铝, 具有不同孔径大小的微孔, 一般平均孔径为(4~7) nm, 新鲜硅酸铝催化剂的比表面积可达(500~700) m2· g-1。
硅酸铝的催化活性来源于其表面的酸性。在有少量水存在时, 由于Al原子的正电性使水分子解离为H+和OH-, 其中OH-与带正电性的Al原子结合, 而H+则在Al原子附近呈游离状态, 即质子酸, 也称B酸。在硅酸铝催化剂的表面, Al、O、Si组成Al— O— Si结构。由于Al— O键趋向正电性较强的Si, 使Al带有正电性, 既非质子酸, 也称L酸。
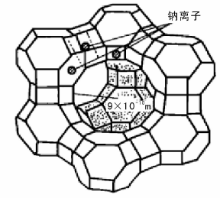
20世纪60年代, 分子筛催化剂在催化裂化中的应用是催化裂化技术的重大发展。与无定型硅酸铝相比, 分子筛催化剂有更高的活性、选择性和稳定性, 因此, 很快就完全取代了无定形硅酸铝催化剂。分子筛也叫泡沸石, 是具有一定晶格结构的铝硅酸盐。由于其规则的晶格结构, 孔径大小均匀, 只能让直径比自己孔径小的分子通过。不同晶格结构的分子筛具有不一样的孔径; 相同晶格结构的分子筛所含金属离子不同时, 孔径也会不同。分子筛按组成和结构的不同可分为A型、X型、Y型及丝光沸石[10]。表4-1列出了工业应用的几种主要的分子筛。目前催化裂化使用的主要是Y型分子筛, 其基本结构为去角八面体, 去角八面体的顶点为Si或Al原子, 之间由O原子相连。而去角八面体又通过O原子连接一半的六边形面形成Y型分子筛的骨架(图4-2)。由八个削角八面体围成的空洞称为八面沸石笼, 它是催化反应进行的主要场所。进入八面沸石笼的主要通道由十二元环组成, 平均直径为(0.8~0.9) nm。钠离子的位置有三处, 其位置如图4-2中所示。
| 表 4-1 几种分子筛的化学组成和孔径 |
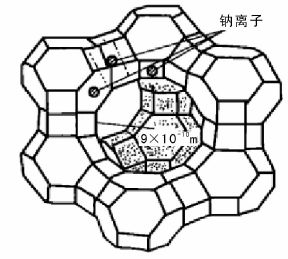 | 图 4-2 Y型分子筛的晶体结构 |
人工合成的分子筛是含钠离子的分子筛, 这种分子筛没有催化活性。分子筛中的钠离子可以用离子交换的方式与其他阳离子进行置换, 用其他阳离子尤其是多价阳离子置换后的Y型分子筛具有很高的催化活性。目前工业上用作催化裂化催化剂的主要是以下4种Y型分子筛:(1) 以稀土金属离子置换得到的稀土-Y型分子筛, 简称REY型分子筛; (2) 以氢离子置换得到的HY型分子筛, 置换方法是先以N置换Na+, 然后加热除去NH3即剩下H+; (3) 兼用氢离子和稀土金属离子置换得到的REHY型分子筛; (4) 由HY型分子筛脱铝得到的有更高硅铝比的超稳Y型分子筛USY。
分子筛也是一种多孔性物质, 具有很大的内表面积, 新鲜分子筛催化剂的比表面积一般为(600~800) m2· g-1。但分子筛是晶体结构, 孔排列规则, 孔直径比较均匀, 其孔径大小为分子大小数量级。研究结果表明, 分子筛催化剂的表面也具有酸性, 由质子酸和非质子酸形成的酸性中心密度比无定型硅酸铝大得多, 其活性也比无定型硅酸铝高得多。当用某些单体烃的裂化速率来比较时, 某些分子筛的催化活性可比硅酸铝高出上万倍。如此高的催化活性在目前的生产工艺中还难以应用, 因此目前工业上所用的分子筛催化剂中仅含有10%~35%分子筛, 其余是起稀释作用的载体和黏结剂。工业上广泛采用的载体是硅酸铝。载体除起稀释作用外, 还有其他重要作用:(1) 在离子交换时, 载体可以容纳分子筛中未除去的钠离子, 从而提高分子筛的稳定性; (2) 在反应和再生时, 载体作为一个热载体起到存储和传递的作用; (3) 增强催化剂的机械强度; (4) 降低催化剂的生产成本。对于重油催化裂化, 载体起着更为重要的作用。重油催化裂化的进料中部分大分子难以直接进入分子筛的微孔中, 如果载体具有适度的催化活性, 则可以使这些大分子先在载体的表面进行适度裂化, 生成较小的分子再进入分子筛的微孔中进一步进行反应。此外, 载体还能容纳进料中易生焦的物质, 如沥青质、重胶质等, 对分子筛起到一定的保护作用。因此, 对于重油催化裂化催化剂, 其载体的活性、表面结构等物理化学性质须认真研究。
分子筛催化剂表面呈酸性, 烃类分子在分子筛上的反应也是按照正碳离子的机理进行。关于分子筛催化剂的活性为何比无定形硅酸铝高得多的问题, 也有许多研究报道。有的研究者认为是由于分子筛上的酸中心密度及酸强度比无定形硅酸铝高得多; 也有研究者认为分子筛晶体结构中存在带正电荷的阳离子和带负电荷的铝氧四面体形成的静电场, 能对被吸附的反应物分子起到极化作用, 从而促进了反应; 还有研究者认为, 分子筛的八面沸石笼中具有较高的反应物浓度, 从而促进了反应。
4.3.2.2 催化裂化催化剂的性能
对一种催化剂而言, 除了需要其化学组成和表面结构数据以外, 还需要一些直接与工业生产相关的指标, 比如活性、稳定性、选择性、密度和机械强度等。
分子筛催化剂的活性在实验室中通常采用微反活性法测定:在微型固定床反应器中放置一定量的待测催化剂(0.5 g), 使用标准原料(轻柴油)在一定温度(460 ℃)、质量空速(16 h-1)和剂油比(3.2)条件下进行反应, 所得产物中汽油(< 240 ℃)、气体和焦炭质量占总进料的百分数即为该催化剂的微反应活性。新鲜催化剂在开始使用的一段时间内, 活性急剧下降, 待降到一定程度后则缓慢下降, 因此初活性并不能真实地反映实际的生产情况。因此在测定新鲜催化剂的活性前先将催化剂进行水热老化处理, 目的就是使测定结果能较接近实际的生产情况。实际生产中, 催化剂受高温和水蒸汽的作用, 其活性逐渐下降; 另一方面, 由于存在催化剂损失需要定期补充新鲜催化剂, 实际生产中催化剂的活性可能维持在一个稳定的水平, 这个活性称为平衡催化剂活性。从生产实际的角度来看, 平衡催化剂活性比新鲜催化剂活性更为重要。平衡催化剂活性的高低取决于催化剂的稳定性和新鲜催化剂的补充量, 对于分子筛催化剂而言一般为60~75。
在一个催化反应中, 人们总是希望催化剂能够有效地促使那些能增加目的产物产率或改善产品质量的反应, 而对其他反应则不起或少起促进作用。如果某种催化剂能够较好地达到这种要求, 那么说明这种催化剂选择性好。对于催化裂化过程, 其主要目的产物是汽油, 如果气体和焦炭产率高, 则气油产率会相应降低, 而高的焦炭产率会增大再生器的负荷。因此催化裂化催化剂的选择性一般以“ 汽油产率/转化率” 以及“ 焦炭产率/转化率” 来表示。裂化催化剂在受到重金属污染后, 选择性会大大变差。重金属污染的程度往往反映在裂化气体中氢含量的增大。因此裂化气中的H2/CH4比值反映了重金属污染的程度, 也反映了催化剂选择性的变化。分子筛催化剂的选择性优于无定型硅酸铝催化剂, 在焦炭产率相同时, 分子筛催化剂的汽油产率高15%~20%。
催化裂化催化剂是多孔性物质, 其密度有几种不同的表示方法:(1) 真实密度:颗粒骨架本身的密度, 又称骨架密度, 一般是(2.0~2.2) g· cm-3; (2) 颗粒密度:即单个催化剂颗粒的密度, 一般是(0.9~1.2) g· cm-3; (3) 堆积密度:催化剂堆积时的密度, 一般是(0.5~0.8) g· cm-3。催化剂的颗粒密度对于催化剂的流化性能有重要影响。
为了避免在生产过程中催化剂过度粉碎以减少损耗和保证良好的流化质量, 要求催化剂具有良好的机械强度, 一般采用“ 磨损指数” 评价微球催化剂的机械强度。测定方法是将一定量的微球催化剂放在特定的仪器中, 用高速气流冲击2 h后, 所生成的< 15 μ m细粉的质量占试样中> 15 μ m催化剂的质量百分数即为磨损指数。通常要求微球催化剂的磨损指数不超过2。
4.3.2.3 工业催化裂化催化剂的种类
按照分子筛分类, 目前工业使用的分子筛催化剂可分为稀土Y(REY)型、稀土氢Y(REHY)型和超稳Y(USY)型。其中REY型分子筛具有裂化活性高、水热稳定性好、汽油收率高的特点, 但其焦炭和干气产率也高, 汽油辛烷值低。主要原因是酸中心多, 氢转移反应能力强。REY型分子筛催化剂一般适用于直馏瓦斯油原料, 采用的反应条件比较温和。REHY型分子筛催化剂是在REY型分子筛催化剂的基础上降低了分子筛中稀土金属离子的交换量, 而以部分H+代替, 使之兼顾了REY型催化剂和HY型分子筛催化剂的优点。REHY型分子筛催化剂的活性和稳定性低于REY型分子筛催化剂, 但通过改性可以大大提高其晶体结构的稳定性。因此, REHY型分子筛催化剂在保持REY型分子筛催化剂较高活性及稳定性的同时, 也改善了催化剂的选择性。REHY型分子筛催化剂中的稀土元素和氢元素的比例可以根据需要进行调节, 从而制成具有不同活性和选择性的催化剂以适应各种需求。USY型分子筛催化剂的活性组分是经过脱铝稳定化处理的Y型分子筛, 这种分子筛骨架有较高的硅铝比、较小的晶胞常数, 其结构稳定性提高、耐热和化学稳定性增强。另外由于脱除了部分骨架中的铝, 酸性中心数目减少, 降低了氢转移反应活性, 使得产物中的烯烃含量增加, 汽油的辛烷值提高, 焦炭产率减少。USY型分子筛催化剂在选择性上具有明显的优越性, 因而发展很快。但是由于其酸性中心数目有所减少, 需要适当提高剂油比来达到原料分子的有效裂化。
裂化催化剂的种类有很多, 根据需要和具体条件选择合适的催化剂, 一般有几个原则:(1) 在渣油的比例增大时, 要选用REHY或者USY型分子筛催化剂。若原料的重金属含量高, 则宜选用具有小比表面积载体的USY型分子筛催化剂; (2) 当要求的产品方案向最大汽油辛烷值方向变化时, 则应当选用REHY或USY型分子筛催化剂; (3) 当再生器的负荷比较紧张时, 应选用焦炭选择性优良的REHY或USY型分子筛催化剂, 当催化剂循环量受到制约时, 宜选用活性高的REHY或REY型分子筛催化剂。
4.3.2.4 催化裂化催化剂的失活与再生
在反应-再生过程中, 裂化催化剂的活性和选择性不断下降, 这种现象称为催化剂的失活, 其原因主要有:高温或与水蒸汽的作用、裂化反应生焦、毒物的毒化。在高温特别是有水蒸汽存在条件下, 裂化催化剂的表面结构发生变化, 比表面积和孔容减少, 分子筛晶体结构遭到破坏, 导致催化剂活性和选择性下降。无定型硅酸铝催化剂的热稳定性较差, 温度高于650 ℃时失活很快。分子筛催化剂的热稳定性比无定型硅酸铝要高很多。REY型分子筛的晶体崩塌温度为(870~880) ℃, USY型分子筛的晶体崩塌温度为(950~980) ℃。实际上当温度高于800 ℃时, 许多分子筛就已经开始发生明显的晶体破坏现象。在工业生产中, 对于分子筛催化剂, 一般在650 ℃以下时催化剂失活很慢, 当温度超过730 ℃时, 失活问题就比较明显。催化反应生成的焦炭沉积在催化剂表面, 覆盖催化剂的活性中心, 使催化剂的活性和选择性下降。随着反应的进行, 催化剂上沉积的焦炭增多, 失活程度也加大。工业催化裂化过程中所产生的焦炭可认为包括4种类型:(1) 催化焦, 即烃类在催化活性中心上反应时生成的焦炭, 其碳氢比较高, 催化焦随反应转化率的增大而增加; (2) 附加焦, 即原料中的焦炭前驱物在催化剂表面吸附, 经缩合反应生成的焦炭, 通常认为在全回炼时附加焦的量与残炭值基本相当; (3) 可汽提焦, 也称剂油比焦, 即由于在汽提段汽提不完全而残留在催化剂上的重质烃类, 碳氢比较高, 数量与汽提段的汽提效率、催化剂的孔结构等因素有关; (4) 污染焦, 即由于重金属沉积在催化剂表面促进脱氢缩合反应而产生的焦, 数量与催化剂上的金属沉积量、沉积金属的类型及催化剂的抗污染能力等因素相关。在实际生产中, 催化剂的毒物主要是某些重金属和碱性含氮化合物。重金属在裂化催化剂上的沉积会降低催化剂的活性和选择性。不同重金属对催化剂的影响方面和程度有所不同, 其中以钒和镍的影响最为重要。在催化裂化条件下, 镍起脱氢催化剂的作用, 使催化剂的选择性变差, 带来的结果是焦炭产率增大、液体产率下降、产品的不饱和度增大、气体中的氢含量增大; 钒会破坏分子筛的晶体结构, 并使催化剂活性下降。在催化剂上的金属含量低于3 000 μ g· g-1时, 镍对选择性的影响为钒的4~5倍, 而在高金属含量时, 钒对选择性的影响与镍达到相同的水平。重金属污染的影响还与其老化程度有关。实践表明, 已经老化的重金属污染作用要比新沉积的重金属作用弱很多。另外重金属污染的影响大小还与催化剂的抗金属污染能力有关。催化剂上的重金属来源于原料油, 国外原油钒含量相对较高, 而我国原油钒含量相对较低。一般情况下, 以瓦斯油为原料时, 重金属污染的程度并不严重, 但是对于来自某些重金属含量很高原油的瓦斯油则必须重视重金属的污染问题。除了重金属外, 碱金属和碱土金属以离子态存在时, 可以吸附在催化剂的酸性中心上并使之中和, 从而降低催化剂活性。实际生产中, 钠离子对催化剂的毒化作用需要注意, 另外, 钠离子会降低催化剂的熔点, 使之在再生温度条件下发生熔化, 破坏分子筛和载体。除了金属毒物以外, 碱性含氮化合物对裂化催化剂也是毒物, 它会使催化剂的活性和选择性降低。碱性含氮化合物毒害作用的大小除了与总含氮量有关外, 还与分子结构有关, 如分子大小、饱和程度等。
裂化催化剂在反应器和再生器之间不断循环, 通常在离开反应器时催化剂上焦炭含量约1%。为了使催化剂能够继续使用, 工业上采用烧去焦炭的方法使其再生。通过再生可以恢复由于结焦而丧失的活性, 但不能恢复由于结构变化以及金属污染而引起的失活。再生是催化裂化的重要过程, 其再生能力对于装置的处理能力至关重要。催化剂上沉积的焦炭主要是缩合反应的产物, 主要成分是碳和氢, 一般情况下氢碳比为0.5~1。催化剂再生反应就是利用空气中的氧烧去沉积的焦炭, 主要反应为:
C+O2→ CO2
Δ H=-33 873 kJ· (kg-C)-1
C+O2→ CO
Δ H=-10 258 kJ· (kg-C)-1
H+O2→ H2O
Δ H=-119 890 kJ· (kg-C)-1
通常氢燃烧的速率比碳快得多, 所以碳的燃烧速率是确定再生能力的决定因素。再生反应是放热反应, 而且热效应相当大, 足以提供装置热平衡需要的热量。
4.3.3 催化裂化工艺流程
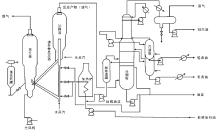
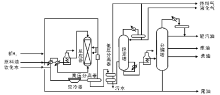
催化裂化装置一般由反应-再生系统、分馏系统、吸收稳定系统和再生烟气能量回收系统组成, 如图4-3所示。
对于提升管催化裂化装置, 原料换热后与回炼油预热喷入提升管反应器的底部, 与高温再生催化剂接触并立即气化。油气携带催化剂沿提升管向上流动, 经快速分离器进入沉降器, 并通过顶部出口进入分馏系统。经快速分离器分出的催化剂由沉降器下部进入汽提段, 用水蒸汽吹去吸附的油气后进入再生器再生, 再生后的催化剂再进入提升管反应器, 完成整个反应-再生循环。由沉降器顶部出来的反应产物进入分馏塔下部, 经分馏后得到富气、粗汽油、轻柴油、重柴油、回炼油和油浆。油浆浓缩后用回炼油稀释送回反应器进行回炼并回收催化剂[11, 12, 13]。
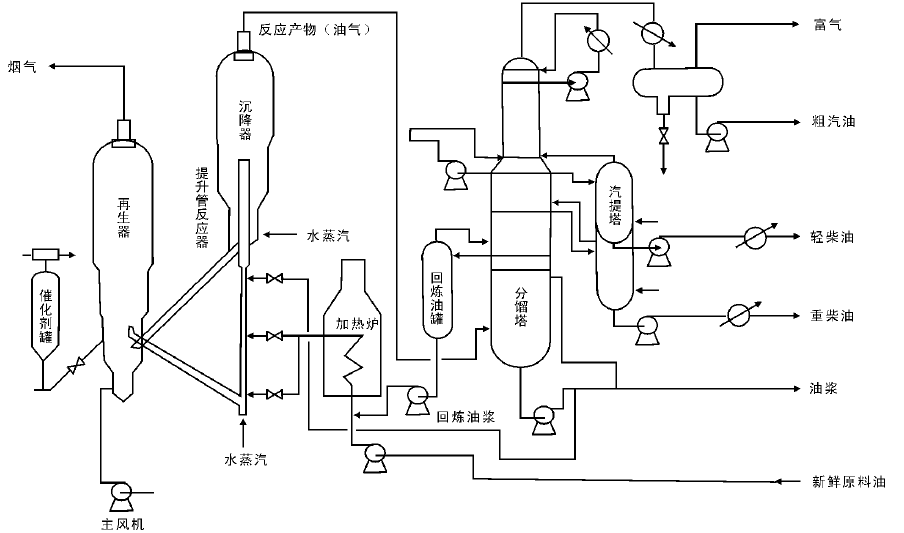 | 图 4-3 催化裂化装置工艺流程 |
4.3.4 加氢裂化反应
目前, 炼油技术中广泛应用于生产轻质和清洁油品的技术仍主要为加氢技术。我国炼油厂加氢能力(包括加氢精制、加氢裂化和加氢处理)约占一次处理能力的30%。相比之下, 美国、德国和日本等技术发达国家的加氢能力占一次处理能力的80%~90%。同时, 在技术发达国家, 加氢裂化工艺约占整个炼油工业二次加工处理能力的13%, 而我国约占5%[14, 15]。这要求我国的炼油结构需要重大调整。国外加氢裂化技术相对成熟, 我国应大力发展以清洁汽油为主的加氢裂化技术, 并以生产化工原料为导向[16]。
加氢裂化过程涉及的反应包括加氢精制反应(加氢脱硫、氮、氧、金属杂质和芳烃)和裂化反应。加氢裂化技术可以同时满足石油加工的轻质化和清洁化要求, 将受到更多炼油行业重视, 也必将发展为炼油的核心技术[1, 17, 18]。在裂化反应中, 根据原料来源, 加氢裂化反应可以分为渣油的加氢裂化、馏分油的加氢裂化和重油的加氢裂化。若根据加氢裂化过程中是否有氢气消耗, 又可以将加氢裂化分为耗氢的裂化反应、不饱和键的加氢饱和反应和不耗氢的异构化反应等。
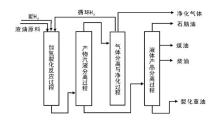
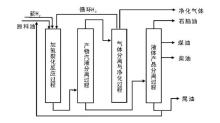
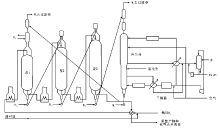
渣油的加氢裂化又可以称为渣油加氢或渣油加氢处理。渣油加氢裂化的原料包括常压重油和减压渣油。在高压和高温条件下, 渣油与氢气在催化剂上反应, 其中的硫、氮化合物分别生成硫化氢、氨气和烃类物质; 金属的有机化合物则生成相应的硫化物和烃类物质; 而渣油主要组成中一些较大的分子则发生裂解并加氢, 转变成较小分子的优质理想组分, 如石脑油和柴油[2]。典型的渣油加氢裂化工艺流程如图4-4所示。
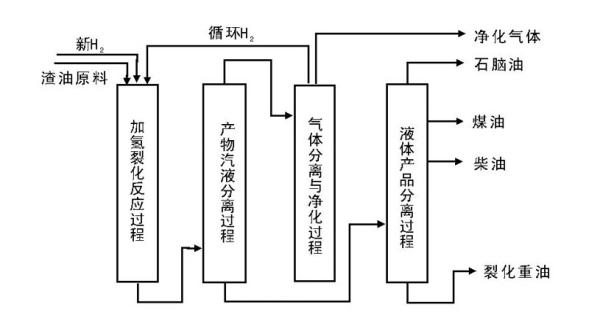 | 图 4-4 典型渣油加氢裂化工艺过程 |
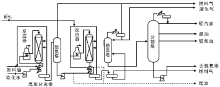
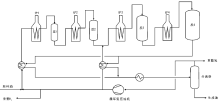
馏分油加氢裂化是将重质馏分油裂化为轻质馏分油的过程, 包括粗汽油加氢裂化生产液化气和减压蜡油、脱沥青油加氢裂化生产航空煤油和柴油。典型的馏分油加氢裂化工艺过程如图4-5所示。
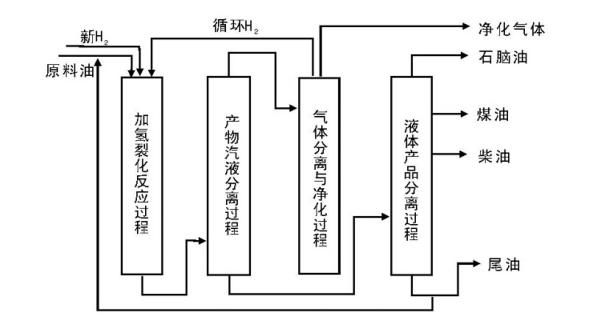 | 图 4-5 典型馏分油加氢裂化工艺过程 |
重油的加氢裂化是指重油在高温、高压和氢气存在条件下, 在催化剂上发生裂化反应, 转化为气体、汽油、煤油、柴油等产品的加氢过程。重油原料主要有减压馏分油、常压重油、减压渣油、脱沥青油等。
烃类的加氢裂化反应是催化裂化反应与加氢裂化反应的组合, 在临氢条件下, 烃类也会发生异构化反应。烷烃的加氢裂化包括C— C键的断裂反应和生成产物中不饱和分子碎片的加氢反应。原料中的烷烃和裂化反应后生成的烷烃均会发生异构化反应。反应中生成的烯烃先进行异构化反应, 之后再加氢生成异构烷烃[19, 20, 21]。烷烃在加氢裂化反应中遵循碳正离子机理, 尤其在具有较强酸性的催化剂上, 该反应机理特征非常明显。由于生成的碳正离子在β 位发生断键, 故气态产品中以C3和C4居多。同时, 烷烃进行加氢裂化后的产品分布和组成取决于烷烃中碳正离子的异构、分解和稳定程度大小。烷烃进行的加氢裂化反应如下所示:
R1 — R2+H2→ R1H+R2H
nCnH2n+2 → iCnH2n+2
以十六烷为例, 其加氢裂化过程如下:
C16H34→ C8H18+C8H16
C8H16+H2 → C8H18
环烷烃在加氢裂化条件下, 主要发生脱烷基、异构化和开环反应。同时, 环烷烃加氢裂化的方向随着催化剂的活性和酸性的不同而改变。单环环烷烃除了异构化、脱烷基侧链和断环反应外, 也会发生不明显的脱氢反应。长侧链的单环六元环烷烃在较强酸性条件下, 多数情况下长侧链会发生断裂, 而六元环很少断裂。短侧链的单环六元环烷烃在较强酸性条件下, 会先异构化生成环戊烷衍生物, 再后续发生其他反应。双环环烷烃在加氢裂化时, 其中一个环先开环并异构化, 生成环戊烷衍生物。随着反应进行, 第二个环也会断开。双环环烷烃的加氢裂化产物中有并环戊烷存在。双环环烷烃的异构化反应包括环的异构化和侧链烷基的异构化。双环环烷烃的加氢裂化也遵循碳正离子机理。总的来说, 环烷碳正离子要比烷烃碳正离子裂化困难, 只有在苛刻条件下, 环烷碳正离子才会发生β 位断裂[22]。环烷烃加氢裂化过程表现出明显的碳正离子机理, 以环己烷和十氢萘[23, 24]的加氢裂化反应为例, 其反应历程如下:
环己烷:
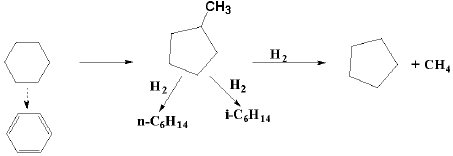
十氢萘:
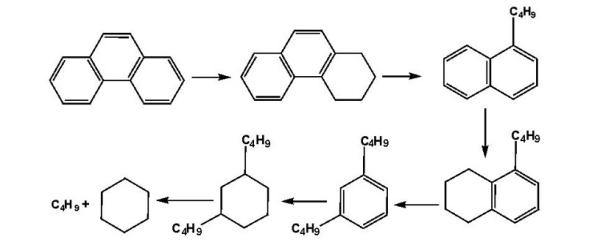
在加氢裂化条件下, 苯先加氢生成环己烷, 之后环己烷会发生上述裂化反应。烷基苯的加氢裂化反应主要包括脱烷基、烷基转移、异构化和环化。在加氢裂化反应中, C1~C4侧链烷基苯以脱烷基反应为主, 生成苯; 其次通过异构化和烷基转移反应, 生成侧链有异构的烷基苯和二烷基苯。具有侧链的烷基苯的裂化反应可以脱烷基生成苯和烷烃, 也可以通过断裂侧链中的C— C键生成烷烃和具有较小短链的烷基苯。侧链较短的烷基苯很难脱烷基, 多发生异构化和歧化反应, 如甲基苯和乙基苯。而侧链较长的烷基苯, 不仅会发生脱烷基、异构化和侧链氢解断裂等反应, 其侧链还可能环化生成双环化合物。在正丁基苯的加氢裂化反应中, 侧链异构反应更容易比脱除侧链反应发生。并且对于脱烷基反应, α -C上的支链越多, 反应越容易发生。以正丁苯的脱烷基为例, 烷基脱除速率依次为:叔丁苯> 仲丁苯> 异丁苯> 正丁苯。苯环上存在侧链烷基使得芳烃加氢困难, 烷基侧链的数目对苯环加氢的影响更甚于侧链长度的影响。
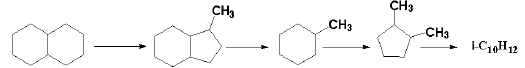
对于多环芳烃, 其在加氢裂化条件下反应复杂, 因为芳烃的芳香环加氢饱和之后才能开环, 开环之后再发生裂化反应[25]。多环芳烃很快加氢生成多环环烷芳烃, 其中环烷环较容易开环, 之后再发生异构化、断侧链和脱烷基等反应。以稠环芳烃为例, 稠环芳烃的加氢过程是逐环加氢、断环, 每个环的加氢和脱氢均处于平衡状态, 并且加氢难度逐环增加。除了加氢裂化反应之外, 在较强酸性下, 稠环芳烃的中间产物也会发生深度异构化、脱烷基侧链和烷基的异构歧化等反应。总的来说, 多环芳烃最终的加氢裂化产物可能以苯类和较小分子的烷烃为主。菲的加氢裂化反应历程如下:
菲:
甲基苯的异构化和歧化反应历程为:
异构化:
歧化:
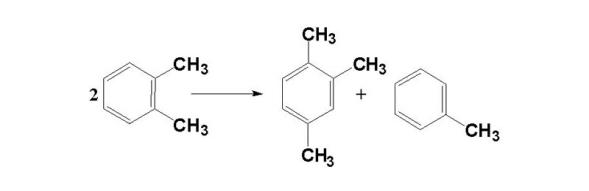
烯烃在加氢条件下主要发生加氢饱和以及异构化反应。这两类反应均可以提高产品的质量。烯烃通过加氢会生成烷烃, 通过异构化反应, 双键位置和烯烃链的空间结构发生变化, 生成的烷烃在加氢裂化条件下后续发生裂化和异构化反应。焦化汽油、焦化柴油和催化裂化柴油在加氢精制条件下, 烯烃的加氢反应很完全, 但并不是关键的加氢反应。烯烃的加氢以及后续异构反应历程为:
nCnH2n+H2→ nCnH2n+2
nCnH2n → iCnH2n
iCnH2n+H2→ iCnH2n+2
4.3.5 加氢裂化催化剂
加氢裂化催化剂是由加氢活性组分和酸性载体组成的典型双功能催化剂, 活性金属组分提供加氢和脱氢功能, 酸性中心提供裂化和异构化功能, 两种功能在催化反应中协同作用, 各司其职。加氢组分主要是Ⅵ B 族和Ⅷ 族金属元素(Ni、Co、Mo 和W)的氧化物、硫化物或者金属(Pt和Pd)。常用的载体是无定形的硅酸铝、硅酸镁和各种分子筛, 近年来主要用各种分子筛作为载体。根据产品的需要, 改变催化剂加氢组分和酸性载体的配比关系, 可以得到不同加氢/脱氢活性和裂化活性的催化剂。一般需要根据原料性质、生产目的等实际情况选择催化剂。在一段加氢裂化工艺流程中, 若要生产中间馏分油, 要求催化剂对多环芳烃有较高的加氢活性, 对原料油中的含氮、硫等有机化合物有较好的抗毒性和中等的裂化活性。而对于两段加氢裂化工艺, 旨在最大限度地生产汽油和中间馏分油, 所用原料比较重并且硫、氮含量高, 其第一段加氢的目的是为第二段加氢裂化制备原料, 此时要求第一段催化剂要有脱硫、脱氮活性; 第二段催化剂必须是由酸性载体制成的具有强的裂化和异构活性的催化剂。当加氢裂化的目的是获取航空煤油时, 则要求催化剂具有较高的脱芳烃活性。对于上述各种催化剂, 都要求催化剂具有较高的稳定性、再生性和抗毒性。
4.3.5.1 加氢裂化催化剂的组成与结构
加氢裂化催化剂的加氢活性组分与加氢精制催化剂活性组分相同, 加氢原理和要求也与加氢精制催化剂相同。一般认为, 活性物种为WS2、MoS2、Pt和Pd等, 而Ni和Co作为助剂。金属或金属硫化物在活性上有差别, 各种金属间匹配也有不同活性, 大致顺序为:贵金属> 过渡金属硫化物> 贵金属硫化物。虽然贵金属加氢和脱氢活性高, 却容易因氮化物存在而中毒, 因此, 在加氢裂化中使用Pt和Pd等贵金属催化剂时, 常需要对原料预进行处理, 预先将氮含量脱除到一定水平。
研究表明, Ⅵ B 族和 Ⅷ 族金属组分相互组合后的活性要比单独组分的加氢活性好得多, 各种组分组合后加氢活性顺序为:Ni-W> Ni-Mo> Co-Mo> Co-W。对于加氢脱氮、加氢脱金属和加氢异构化反应, 上述加氢活性顺序不变, 但是对于加氢脱硫反应, Co-Mo加氢活性则最高。活性组分间的组合也存在最佳原子比, 在最佳原子比下, 催化剂可以得到最好的加氢脱氮、加氢脱硫、加氢裂化和异构化活性。
对于不同的加氢裂化催化剂, 其酸性组分、加氢活性组分最佳原子比不一定相同。催化剂中各个活性组分如何搭配组合, 不仅要考虑加氢组分, 也要考虑最佳原子比以及催化剂整体的制备成本。
催化剂的裂化功能主要由酸中心提供, 一般认为裂化反应通过酸催化实现。而裂化性能则取决于催化剂本身的性质如酸量、酸强度和比表面积。加氢裂化催化剂的载体有酸性和弱酸性两种。酸性载体为硅酸铝、硅酸镁、分子筛等; 弱酸性载体为氧化铝和活性炭等。酸性载体可以提供酸中心, 提高催化剂的机械强度、热稳定性和抗毒性能, 增加催化剂比表面积并提供合适的孔隙结构, 促进活性组分分散以及减少组分用量, 降低成本。
4.3.5.2 加氢裂化催化剂的性能
以分子筛为载体的加氢裂化催化剂的反应活性高, 灵活性大, 寿命长; 以无定形硅铝为载体的加氢裂化催化剂, 虽然活性低, 灵活性差, 但产品质量稳定, 中油选择性高。因此, 初期的高中油型的加氢裂化催化剂多以无定形硅铝为载体。
20世纪60年代中期, 工业上逐渐采用分子筛作为加氢裂化催化剂的载体。含分子筛的加氢裂化催化剂酸中心的强度和类型与无定形硅酸铝类似, 但是酸中心的数量却是无定形硅酸铝的10倍, 并且酸性可以通过广泛调节阳离子组成以及骨架硅铝比控制。作为载体的分子筛结构类型和阳离子类型可以不尽相同, 而且还可以用不同的方法添加分子筛于催化剂中。这样, 通过有目的调节催化剂活性和选择性, 就可以制备不同原料性质和生产目的的催化剂。工业使用的分子筛包括Y型和超稳Y型, 还有毛沸石、丝光沸石、凌钾沸石和ZSM系列, 还可以将分子筛经过不同处理来满足不同要求(如 HY、USY、HPY、UHPY、REY等)。近些年, 又有一种SSY分子筛引起人们的关注, 这类分子筛抗氮能力和酸强度均明显高于超稳Y型分子筛。
随着对分子筛结构和改性技术研究的不断深入, 目前主流的中油型、高中油型的加氢裂化催化剂载体, 主要由无定形硅铝和(或)沸石分子筛组成, 沸石分子筛主要是各种改性Y、β 以及Y与β 及其他分子筛的复合分子筛。分子筛的改性处理, 提高了分子筛的稳定性, 降低了分子筛的酸中心数, 形成了一些 (5~20) nm的二次孔, 提高了分子筛对大分子烃的转化能力, 加快了反应物和产物的扩散速率, 减少了二次裂化反应的发生, 提高了催化剂的中油选择性。使用上述改性处理Y分子筛为载体主要组分的加氢裂化催化剂的性能, 已经达到以无定形硅铝为载体的高中油型加氢裂化催化剂的性能。近几年, 高中油型加氢裂化催化剂不仅致力于Y、β 分子筛的改性研究, 也引入了ZSM、SAPO、MAPSO、MeAPO、MCM系列分子筛, 为加氢裂化催化载体提供了更为广阔的材料来源。尤其是介孔分子筛的出现, 介孔分子筛具有高度有序的纳米孔径[(2~40) nm]、超高的比表面积(约1 500 m2· g-1)。利用介孔分子筛作为加氢裂化催化剂载体, 用于大分子烃类的加氢裂化催化转化过程, 可以从本质上提高加氢裂化催化剂催化性能。
目前, 催化剂载体研究的重点集中于催化剂改性和合成方法调整上, 以求得到高活性、适当酸性和高稳定性的分子筛。同时, 一些分子筛已经实现工业化应用, 这些分子筛由于不同的结构特征和酸性能, 成为不同反应体系实现最优化操作的选择。
4.3.5.3 工业加氢裂化催化剂的种类
加氢裂化催化剂作为加氢裂化技术的核心, 开发和研制一直受到国内外各大研究机构及石油公司的高度重视。加氢裂化催化剂按目的产品可分为轻油型(以最大量生产石脑油和部分中间馏分油为目的产品)、中油型(以生产中间馏分油和部分石脑油为目的产品)、高中油型(以最大量生产中间馏分油, 即煤油和柴油为目的产品)和重油型(以生产润滑油基础油原料为目的产品)4种。近几年, 为实现最大量生产优质中间馏分油的目的, 加氢裂化催化剂的一个重要开发方向就是开发加氢裂化多产中间馏分油催化剂。
国外从事加氢裂化催化剂研制开发并且催化剂性能处于领先水平的研究机构, 主要有Chevron、UOP、Mobil、Criterion、Shell、Akzo Nobel和IFP公司, 其中, UOP、Chevron和Criterion公司的加氢裂化催化剂约占全世界加氢裂化装置使用的加氢裂化催化剂总量的90%。UOP公司比较有代表性的最大量生产中间馏分油的非贵金属加氢裂化催化剂有DHC-20、DHC-39、DHC-41和DHC-110; Chevron公司灵活生产中间馏分油-润滑油料或乙烯料的非贵金属加氢裂化催化剂有ICR-126、ICR-147和ICR-150; Criterion公司生产最大量中间馏分油的加氢裂化催化剂有Z-503、Z-603、Z-623和Z-723。目前全世界采用UOP技术的加氢裂化装置已超过140套, 装置加工量约占总加工量的80%。因此, UOP加氢裂化催化剂的发展现状和水平具有较大的代表性[26, 27, 28, 29]。DHC-39催化剂曾在金陵石化公司加氢裂化装置上应用, 在> 350 ℃馏分油转化率是70%和80%两种转化深度下, 加工伊朗VGO, 反应温度为377 ℃和386 ℃, 中油选择性80.3%和75.5%, (132~370) ℃中间馏分油的收率为55.7%和61.2%[30]。我国在中油型、高中油型加氢裂化催化剂方面的开发研制工作主要以中国石化抚顺石油化工研究院为主, 开发研制出了3901、3974、FC-16和FC-20 等最大量生产中间馏分油的加氢裂化催化剂, 并达到国际先进水平。FC-16催化剂处理大庆VGO在> 350 ℃馏分油转化率为80%转化深度下, 反应温度为368 ℃, 中油选择性80.7%, (132~370) ℃中间馏分油收率为64.0%[31]。
4.3.5.4 加氢裂化催化剂的制备
加氢裂化催化剂也是负载型催化剂, 制备方法与加氢精制催化剂相似, 可以参考加氢精制催化剂制备方法, 此处不再赘述。但值得强调的一点是加氢裂化催化剂在制备过程中必须加入较多的酸性载体组分(20%~60%), 如分子筛等裂化活性组分。所采用的分子筛必须经过改性如与H+、N或者稀土离子等阳离子交换获得较强的酸性, 还要经过稀酸或者高温水蒸汽进行扩孔处理, 可以产生更多的二次孔, 这样有利于大分子的扩散和裂化反应, 提高活性和稳定性。
4.3.6 加氢裂化工艺流程
在美国已经工业化的加氢裂化工艺包括:埃索麦克斯(Isomax)、联合加氢裂化(Unicracking/JHC)、H-G加氢裂化(H-G hydrocracking)、超加氢裂化(Ultra-cracking)、壳牌公司的加氢裂化(Shell)和BASF-IFP 加氢裂化。这几种工艺中, 超加氢裂化、H-G加氢裂化和壳牌公司的加氢裂化主要用于生产重整原料(汽油馏分), 而其他几种工艺既可以生产重整原料, 也可以生产航空煤油和柴油。国外其他加氢裂化技术还包括CLG公司加氢裂化技术、UOP公司加氢裂化技术、Haldor Topose公司加氢裂化技术、Albemarle 公司加氢裂化技术和Axens 公司加氢裂化技术。国内加氢裂化工艺主要有中国石化抚顺石油化工研究院工艺和中国石化石油化工科学研究院工艺。
各种加氢裂化工艺的实际流程差别不大, 主要取决于采用的催化剂类型和种类。所用催化剂不同, 工艺条件、产品分布和产品质量也不同。根据原料性质、产品要求和处理量大小, 加氢裂化装置基本上按照一段法和两段法两种流程操作。一段加氢裂化流程也包括两个反应器串联在一起的裂化流程。一段加氢裂化流程用于由粗汽油生产液化气, 或由减压蜡油、脱沥青油生产航空煤油和柴油。两段加氢裂化流程中, 原料首先在第一段(精制段)用加氢活性高的催化剂进行预处理, 经过加氢精制处理的生成油再作为第二段的原料油进料, 在裂解活性较高的催化剂上进行裂化反应和异构化反应, 最大限度地生产重整原料或者中间馏分油。两段加氢裂化流程操作对原料的适用性广, 操作灵活性强, 可用于处理催化裂化循环油、高硫与高氮减压蜡油、焦化蜡油以及这些油的混合油, 亦适合处理一段加氢裂化难处理或不能处理的原料和劣质渣油, 能够最大限度生产汽油或中间馏分油。
如图4-6所示, 以大庆直馏蜡油馏分[(330~490) ℃]的一段加氢裂化流程为例, 原料油经泵升压至16 MPa后与新H2和循环H2混合, 再与约420 ℃的加氢生成油换热至(320~360) ℃进入加热炉。反应器进料温度为(370~450) ℃, 原料油在反应温度(380~440) ℃、空速为1.0 h-1、氢油体积比为2 500的条件下进行反应。最终可以得到燃料气、液化气、轻汽油、航空煤油、低凝柴油和尾油(塔底油)。尾油可部分或全部用作循环油, 与原料油混合再去反应。一段加氢裂化工艺可以有3种操作方案:原料一次通过、尾油部分循环和尾油全部循环。
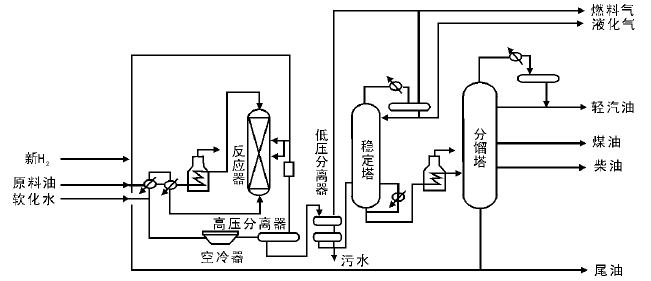 | 图 4-6 一段加氢裂化工艺流程 |
图4-7为两段加氢裂化工艺流程。原料油经泵升压并与新H2和循环H2混合后首先与生成油换热, 再在加热炉中加热至反应温度, 进入第一段加氢精制反应器, 在具有高加氢活性的催化剂上进行脱硫、脱氮和脱金属反应。反应生成油经过换热进入高压分离器分离出循环H2。生成油进入脱硫(氨)塔, 脱去氨气和硫化氢, 再作为第二段加氢裂化反应器的进料。第二段进料与循环H2混合后, 进入第二段加热炉, 加热至反应温度, 在装填有高酸性催化剂的第二段加氢裂化反应器中进行裂化反应。反应物经换热和分离, 分出溶解气和循环H2后送至稳定分馏系统。两段加氢裂化工艺可以有两种操作方案:(1) 先第一段精制, 第二段再加氢裂化; (2) 除加氢精制以外, 第一段还要进行部分加氢裂化, 第二段之后再加氢裂化。后者的特点在于第一段反应生成油和第二段生成油一起进入稳定分馏系统, 分出的尾油可以作为第二段的进料。而且采取第二种方案, 由于裂化深度较大, 汽油、煤油和柴油的收率均增加, 尾油收率明显降低。但试验表明, 若就产品的主要性能, 两种方案无明显差别。
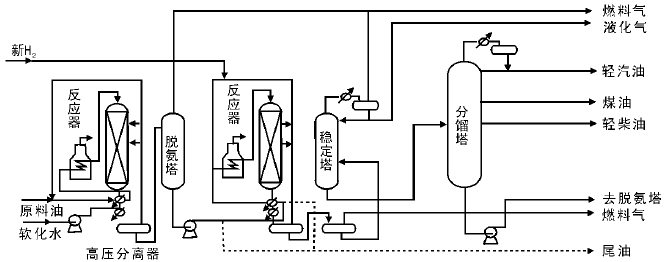 | 图 4-7 两段加氢裂化工艺流程 |
串联加氢裂化工艺中有两个反应器串联, 其他部分与一段加氢裂化工艺均相同, 其中, 对于串联的两个反应器, 分别填装不同的催化剂, 第一个反应器中装入具有高的加氢活性的催化剂, 进行脱硫、脱氮和脱金属反应, 第二个反应器装入抗氨气和抗硫化氢的分子筛型加氢裂化催化剂, 如图4-8所示。
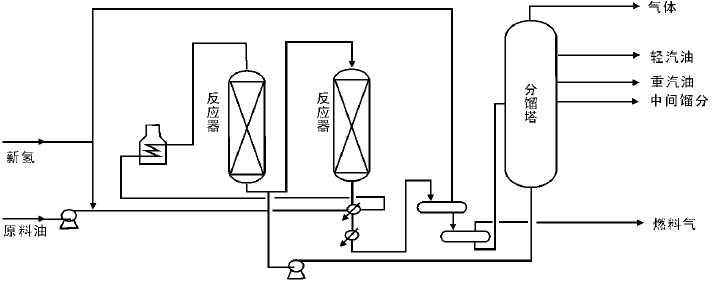 | 图 4-8 串联加氢裂化工艺流程 |
与一段加氢裂化相比, 串联加氢裂化工艺只通过改变操作条件, 就可以最大限度地生产重整原油、航空煤油和柴油。如降低第二个反应器的温度就可以多生产航空煤油或者柴油; 反之, 若升高第二个反应器的温度, 则多生产出重整原油。
同一种原料分别用上述3种加氢裂化工艺处理原料油, 试验结果表明, 从生产航空煤油方面看, 一段加氢裂化工艺航空煤油收率最高, 但重整原料收率低; 从流程结构和投资方面看, 一段加氢裂化工艺优于其他工艺流程; 串联加氢裂化工艺在生产重整原油方面很灵活, 但航空煤油收率偏低。总体上, 两段加氢裂化工艺灵活性最大, 航空煤油收率高, 还能生产重整原料油。两段加氢裂化工艺和串联加氢裂化工艺对原油的质量要求不高, 可以处理高密度、高干点、高含硫、含氮以及高残炭值的原料油。相比之下, 一段加氢裂化工艺对原料油质量要求更严格。国外炼油厂多认为两段加氢裂化工艺最好, 既可以处理一段加氢裂化工艺不能处理的原料油, 又能生产优质的航空煤油和柴油。在投资上, 两段加氢裂化工艺成本略高于一段一次通过加氢裂化工艺, 略低于一段全循环加氢裂化工艺。目前, 许多国家多用两段加氢裂化工艺处理重质原料油生产重整原料油, 以扩大芳烃的来源。
加氢裂化工艺的应用范围日益扩大, 但是用于汽油时辛烷值损失大, 故提升管催化裂化技术生产汽油逐渐取代加氢裂化生产汽油。加氢裂化技术不断发展, 其催化剂仍然在不断改进, 并且该技术正朝低压、低能耗方向发展。临氢降凝技术也属于加氢裂化一部分, 它是在H2和催化剂共存的条件下链烷烃选择性裂化, 也成为加氢脱蜡技术。这种脱蜡技术越来越多地用于降低柴油或者润滑油的凝点, 其催化剂多为负载有活性金属的择形沸石分子筛。
4.4 加氢精制中的催化作用
加氢精制属于石油加工的一个重要过程, 在现代石油化工中不可缺少[32, 33], 是炼油厂目前采用的主要加氢过程。加氢精制主要对油品进行精制, 除去油品中的杂原子(S, N, O)以及金属杂质(主要是Ni和V), 并通过加氢反应减少烯烃含量和部分芳
烃含量, 改善油品质量, 提高轻质油收率, 改善原料来源结构和使用性能。通常情况下渣油加氢裂化后的产品也需要加氢精制。
加氢精制反应主要包括加氢脱硫、加氢脱氮、加氢脱氧、加氢脱金属和加氢脱芳烃反应[1, 2]。在加氢脱硫过程中, 也伴随加氢脱氮、加氢脱氧、加氢脱金属和加氢脱芳烃的反应, 氮、氧化合物中的氮、氧原子分别以NH3和H2O的形式脱除, 而金属化合物(主要是含Ni、V的化合物)中的金属则以硫化物的形式沉积在催化剂的表面。
4.4.1 加氢精制中的化学反应
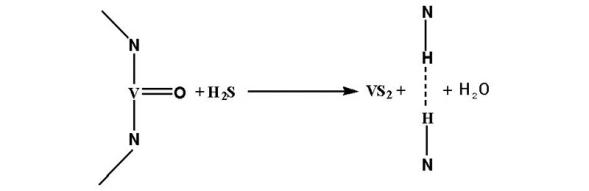
4.4.1.1 加氢脱硫反应
加氢脱硫是指通过催化加氢反应, 使石油组分中的含硫化合物发生氢解而转化成相应的烃和H2S, 从而脱除硫原子。其基本的化学反应包括C— S键的断裂和断裂物的饱和。加氢脱硫反应一般在高温和高压条件下进行, 工业上加氢脱硫反应温度通常为(250~350) ℃, 压力为(2~10) MPa。石油馏分中的含硫化合物有硫醇、硫醚、二硫化物、噻吩、苯并噻吩和二苯并噻吩及其衍生物。由于各种油品中所含有的含硫化合物分子结构和大小不同, 其脱硫机理也有所不同, 在加氢脱硫反应中的活性也不同。传统加氢催化剂上活性大小按以下顺序减小:硫醇> 二硫化物> 硫醚> 噻吩> 苯并噻吩> 二苯并噻吩> 烷基取代的二苯并噻吩[34]。
加氢脱硫反应条件下, 脱硫过程如下[35, 36, 37]:
硫醇:
RSH+H2→ RH+H2S
硫醚:
RSR’ +H2→ R’ SH+RH
R’ SH+H2→ R’ H+H2S
二硫化物:
RSSR’ +H2→ R’ SH+RSH→ R’ SR+H2S
R’ SR+2H2→ R’ H+H2S+RH
噻吩:
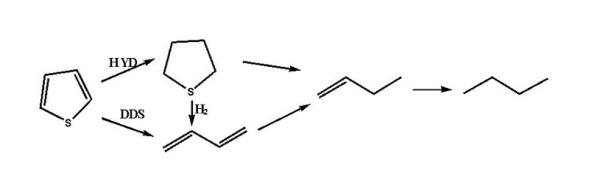
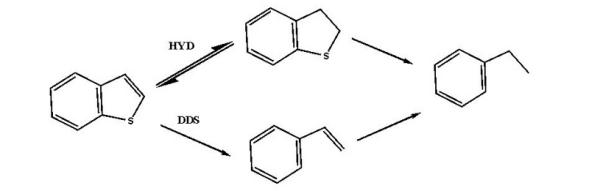
苯并噻吩:
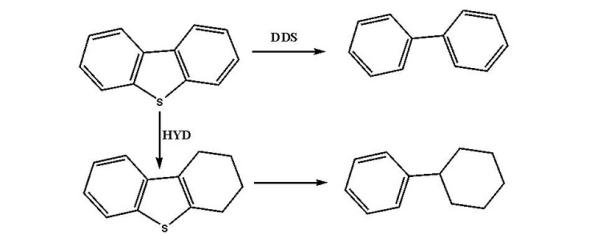
二苯并噻吩:
4, 6-二甲基二苯并噻吩:
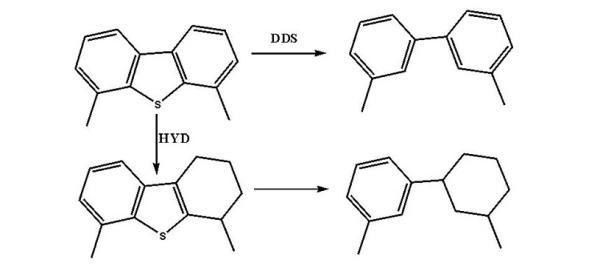
噻吩类化合物的加氢脱硫反应主要有加氢途径和直接脱硫途径。其中反应温度、氢分压、氢油比和产生的H2S均对反应物的转化率有影响。在一定温度范围内, 温度升高, 转化率增大; 氢气压力增大, 反应转化率升高; 增大氢油比有利于脱硫反应, 但当其增大至一定程度, 脱硫率几乎不变。而反应体系中产生的H2S对加氢途径和直接脱硫途径均有抑制作用, 故反应体系中一般维持微量的H2S分压, 用以确保催化剂使用寿命。除此之外, 原料油中其他的含氮、含氧物质、烯烃和芳烃均会与脱硫反应形成竞争反应, 而且在不同程度上均会抑制脱硫反应。在实验室条件下, 苯并噻吩和4, 6-二甲基二苯并噻吩的动力学反应均为准一级反应[38, 39]。
4.4.1.2 加氢脱氮反应
加氢脱氮反应是重油和渣油深度加工的重要工艺。加氢脱氮反应与加氢脱硫反应过程不同, 由于杂环中C═N键的断裂能是杂环中C— N的两倍, 故芳香杂环化合物的加氢脱氮必须先经过芳香环的加氢饱和反应, 才能进一步脱氮。加氢脱氮反应比加氢脱硫要求更高的反应温度和压力。
石油馏分中的有机含氮化合物主要分为非杂环和杂环化合物两类。非杂环化合物包括脂肪胺、苯胺和腈类化合物, 杂环化合物又分为碱性和非碱性杂环化合物。非碱性杂环化合物包括吡咯、吲哚和咔唑等五元杂环, 碱性杂环化合物包括吡啶、喹啉、异喹啉、吖啶、菲啶和苯并喹啉等六元杂环。
由于竞争吸附, 碱性氮化合物的脱氮速率通常较非碱性氮化合物快。在所有有机含氮化合物中, 脂肪胺的反应活性最强。对于催化剂CoMo/Al2O3, 加氢脱氮活性顺序为:吡咯> 吲哚> 吡啶> 喹啉> 苯胺。而在催化剂NiW/Al2O3上, 活性大小顺序为:烷基胺=苯胺> 吲哚> 喹啉。一般认为含氮化合物的脱氮活性顺序为:吲哚> 甲基化苯胺> 单甲基取代吲哚> 喹啉> 咔唑> 甲基化咔唑[40]。加氢脱氮反应条件下, 脱氮过程如下[41, 42, 43, 44, 45]:
烷基胺:
R— CH2NH2+H2→ RCH3+NH3
吡咯:
吲哚:
吡啶:
喹啉:
吖啶:
咔唑:
含氮化合物的相对加氢反应速率与化合物种类、操作条件(温度、压力、氢油比)和催化剂种类有关。在一定温度范围, 温度升高, 脱氮率增大; 氢气分压增大, 脱氮率升高; 增大氢油比也有利于脱硫反应, 但当其增大至一定程度, 脱氮率几乎不变。在大多数杂环含氮化合物的加氢脱氮反应中, 脱氮一般经历两步, 即杂环加氢和C— N键氢解。一般认为喹啉的加氢脱氮反应符合一级反应动力学模型。
4.4.1.3 加氢脱氧反应
大多数石油的氧含量为0.1%~1.0%(质量分数)。石油中的含氧化合物分为酸性氧化物和中性氧化物。酸性氧化物又称石油酸, 包括羧酸(如环烷酸, 脂肪酸和芳香酸)和酚类。中性氧化物包括酮类、酯类、醚类和呋喃类。中性氧化物在石油中含量极少, 故石油中的含氧化合物以酸性氧化物为主。
各种含氧化合物的加氢反应主要包括环的加氢饱和以及C— O键的氢解。含氧化合物的加氢历程如下[46]:
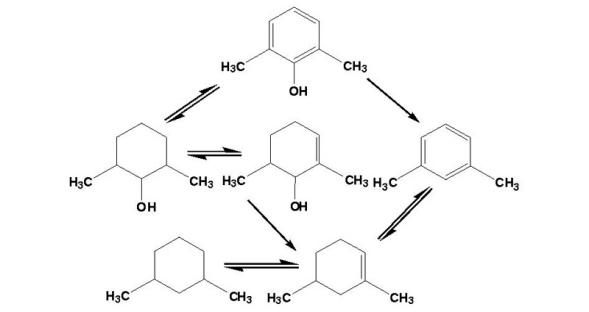
环烷酸:
酚类:
呋喃:
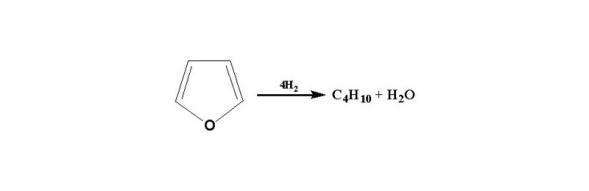
烷基取代酚类:
苯并呋喃:
4.4.1.4 加氢脱金属反应
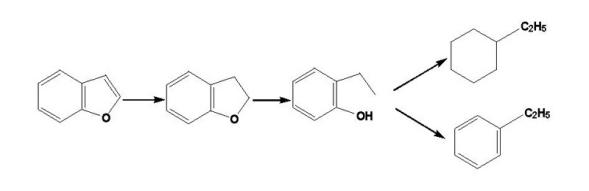
原油重质化和劣质化程度加深, 在渣油加氢方面, 加氢脱金属问题日益突出。渣油中的金属分别以卟啉化合物和非卟啉化合物两种形式存在(如环烷酸铁、钙、镍等), 其中, 油溶性的金属环烷酸盐反应活性很高, 易以硫化物形式沉积于催化剂孔口, 堵塞孔道。而渣油中主要的金属杂质镍和钒以宽范围分子量分布的卟啉形式存在。在不同实验条件下, 如不同的渣油原料、不同类型的反应器和催化剂, 渣油的脱镍和脱钒反应动力学不尽相同, 它们的反应级数在0.5~2之间[47]。
在加氢条件下, 渣油中的金属脱除反应历程为:
脱钒:
脱镍:
4.4.1.5 加氢脱芳烃反应
石油中的芳烃包括单环芳烃(如苯)、稠环芳烃(如萘、蒽、菲)和多环芳烃(联苯、联多苯、多苯代脂肪烃)。柴油中所含的芳烃使得柴油发动机排放亚微米级物质PM(包括碳微粒和可溶性有机物质), PM会引起一些疾病, 存在潜在致癌危险[48]。当前发达国家已对油品中芳烃含量做了限制[49]。通过加氢饱和脱除芳烃可以降低催化裂化和加氢裂化原料的生焦量, 从而提升油品质量和使用性能。
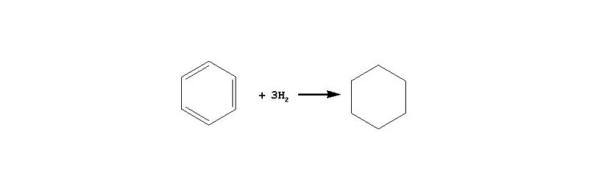
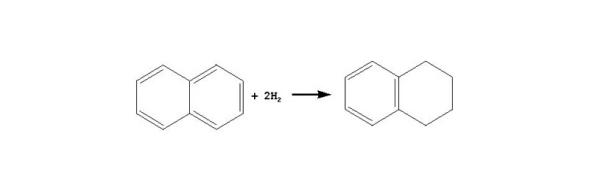
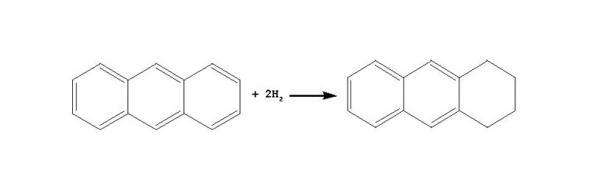
在加氢条件下, 苯先加氢生成环己烷, 环烷烃之后发生裂化。一般情况下, 芳烃加氢反应的平衡常数Kp随温度升高而降低; 芳烃中芳香环数目越多, Kp下降越快。稠环芳烃中第一个环加氢的Kp较大、第二个环加氢的Kp次之, 环越多, 环加氢的Kp越小。从热力学角度看, 稠环加氢的途径主要是先对一个芳香环加氢生成环烷环, 环烷环断裂或者异构, 第二个芳香环再加氢, 后续加氢反应如此继续进行。
芳烃第一个芳香环加氢历程如下所示:
苯:
萘:
蒽:
菲可能的加氢和裂化反应历程为:
4.4.2 加氢精制催化剂
4.4.2.1 加氢精制催化剂的组成和性能
加氢精制催化剂的活性组分是催化剂活性的来源, 活性组分可以是贵金属或者是非贵金属。非贵金属主要来自Ⅵ B 族和Ⅷ 族, 包括 Ni、Co、Mo 和W; 贵金属组分包括 Pt和 Pd。催化剂的加氢活性与其吸附特性有关, 而吸附特性又与催化剂的几何特性以及电子特性有关。根据“ 多位学说理论” — — 凡是适合作为加氢催化剂的金属, 都应具有立方晶格或者六角晶格和“ 半导体理论” — — 具有未填充满的d电子层的金属元素具有催化活性, Ni、Fe、Co、Mo、W、Cr、V、Pd 和 Pt都属于d电子层未填充满的金属元素, 而且均同时具有体心或面心立方晶格或六角晶格, 故均可以用作加氢催化剂的活性组分。
在工业催化剂中, 常常配合不同的活性组分使用来达到加氢活性效果最优化。目前, 工业上常用的加氢催化剂以Mo和W为主催化剂, Ni和Co作为助剂, 氧化铝为载体。在一定范围内, 活性金属含量越高, 加氢活性越高。综合生产成本和催化剂活性增加幅度, 以金属氧化物含量质量百分数计, 目前加氢精制催化剂活性组分含量一般为15%~35%。活性组分的组合包括Co-Mo、Ni-Mo、Co-W和Ni-W等, 不同的活性组分组合对各类反应活性顺序为:
加氢脱硫:Co-Mo> Ni-Mo> Ni-W> Co-W;
加氢脱氮:Ni-W> Ni-Mo> Co-Mo> Co-W;
加氢脱氧:Ni-W≈ Ni-Mo> Co-Mo> Co-W;
加氢脱烯烃:Ni-W> Ni-Mo> Co-Mo> Co-W。
最常用的加氢脱硫催化剂是Co-Mo型, 而对于含氮较多的原料则需要选择Ni-Mo或者Ni-W型加氢精制催化剂。继二元活性组分组合基础之后, 系列三元活性组分甚至四元活性组分组合型加氢精制催化剂也相继被开发出来, 如Ni-Co-Mo、Ni-Co-W、Ni-Mo-W和Ni-Co-Mo -W催化剂。这些催化剂兼具有加氢脱硫、加氢脱氮和加氢脱芳烃等优异的加氢性能。
添加助剂于加氢精制催化剂可以改善某方面性能, 包括选择性、活性和稳定性。大多数助剂为金属助剂, 也有少量的非金属助剂。助剂作用于催化剂的机理不全相同, 有的是结构型助剂, 有的是调变型助剂。前者可以增大催化剂比表面积, 减缓烧结并提高催化剂的稳定性, 如K、Ba、La的添加可提高催化剂抗烧结性能。后者作为调变助剂, 可以通过增加或者减少未充满的d轨道电子数目、改变活性组分中原子间距离或者价带宽度以及钝化副反应的活性中心来调变催化剂的电子结构、表面性质或者晶型结构, 从而改变催化剂活性和选择性。助剂本身活性并不高, 但是与主要活性金属组分以合适比例组合后制备的催化剂则具有优异的活性。
为了提高催化剂活性组分的分散性, 并提高催化剂的加氢脱氮性能和芳烃饱和性能, 也会加一些助剂如P、B、F、Si 和Ti等。加入少量的P、F等酸性组分, 既可以促进活性组分的分散, 增加金属的利用率, 还可以提高C— N键的裂化活性和芳烃饱和活性。
加氢精制催化剂的载体有中性载体和酸性载体两大类。中性载体有活性氧化铝、活性炭和硅藻土等, 酸性载体有硅酸铝、硅酸镁和分子筛等。载体一般没有活性, 但由于其大的比表面积可用于分散活性组分, 减少活性金属使用量。而且载体也可以作为催化剂的骨架, 进而提高催化剂的稳定性和机械强度。载体也可以保证催化剂具有一定的形状和大小, 使之符合工业反应器中流体力学条件的需要, 减少流体的阻力。载体还可以与活性组分相配合而使活性、选择性以及稳定性发生变化。
除了加氢催化剂的化学组成影响活性外, 催化剂的物理性质如比表面积、孔容、孔径分布、颗粒度以及催化剂外形都会影响活性组分作用的发挥。在重质油的加氢精制过程中, 常存在床层压降和扩散阻力, 为了解决这些问题, 常将催化剂制备成三叶草、四叶草和辐条形状, 直径约1.5 mm。而且随着加氢精制原料变重, 原料分子变大, 对加氢精制催化剂的孔径分布有一定要求。研究发现, 重质油加氢脱金属反应是扩散控制, 要求催化剂的孔道结构为双峰孔分布, 较合适的孔径范围为(15~25) nm; 并且采用双峰孔催化剂使引入的大孔增加了分子扩散的通道, 显著增加催化剂脱金属活性。对于渣油的加氢脱硫反应, 不仅要考虑扩散的影响, 也要考虑催化剂加氢活性。对于脱氮反应, 即要求催化剂有非常强的加氢能力, 也要求催化剂有高的比表面积。
国内外各大石油公司多数均有自己的加氢精制催化剂。由于原料不同和生产目的不同, 加氢精制催化剂品种繁多。
4.4.2.2 加氢精制催化剂的制备
在柴油加氢精制催化剂方面, 2001年由雅宝、埃克森和日本凯金3家公司开发了非负载的体相加氢催化剂— — Nebula。Nebula催化剂用于处理重油和渣油均显示出优异的加氢性能, 研究者又改良升级Nebula催化剂并研发了Nebula-20催化剂。目前, Nebula系列催化剂受到人们广泛关注, 逐渐成为加氢领域研究热点。工业应用的石油馏分加氢精制催化剂一般是负载型催化剂, 即将活性组分Ni、Co、Mo和W的氧化物负载于多孔载体上制备成催化剂。对于负载型催化剂的制备, 常规方法是浸渍法和混捏法。浸渍法是将活性组分浸渍到载体上, 主要制备步骤是:(1) 先将载体粉料(氧化铝干胶粉)与一定量的助剂(如分子筛)、扩孔剂、助挤剂(田箐粉)和粘结剂(硝酸水溶液)混合, 经充分混捏后在挤条机上挤出成型, 然后经过干燥(烘干)、焙烧[(300~600) ℃、(4~8) h]制成催化剂载体; (2) 将加氢活性组分前驱体(采用易分解很少有元素残留的金属盐类, 如Ni和Co的硝酸盐、碳酸盐以及醋酸盐, Mo和W的铵盐、氧化物和对应的酸, Pt和Pd常用氯铂酸和氯化钯)的溶液浸渍成型后的载体, 再干燥和焙烧制成催化剂。
浸渍过程可以采用分步浸渍或者共浸渍, 目前共浸渍方法较为普遍。因为共浸渍法制备步骤简单, 节约制备成本和时间, 有助于发挥助催化剂Ni、Co与主催化剂Mo、W的协同效应。但缺点在于Ni、Co与Mo、W的混合溶液稳定性差, 可配区域范围窄, 必须仔细控制各组分配比、加入顺序和溶液的pH, 并且还需要加入适当的稳定剂(如磷酸、氨水、柠檬酸、酒石酸等, 而且加入的有机酸类稳定剂有助于活性组分的分散)。分步浸渍是先浸渍Mo或W, 再浸渍Co或Ni, 每次浸渍后均经过干燥和焙烧, 这样可以使催化剂活性达到最优化。
在实际工业生产中, 浸渍法又分为饱和浸渍法和过饱和浸渍法。饱和浸渍法也称等体积浸渍法, 是指活性组分浸渍液的体积等于催化剂载体能够吸收的浸渍液的最大体积(饱和吸附量), 这样, 浸渍液中的活性组分全部浸渍到载体上, 可以比较容易地控制活性金属的负载量。饱和浸渍法比较适合生产小批量的加氢精制催化剂以及制备负载型贵金属催化剂。过饱和浸渍法是指活性组分浸渍液的体积大于催化剂载体能够吸附的浸渍液的最大体积(饱和吸附量), 因此, 浸渍液中的活性组分部分浸渍到载体上。此法不足之处在于活性组分的负载量较难控制, 这是因为不同组分竞争吸附力不同。而且过饱和浸渍法浸渍后需要除去多余浸渍液, 若要再次使用沥出的浸渍液, 需要重新测定其中各个活性组分含量重新调配, 操作比较繁琐费时。但优点在于催化剂上活性组分的负载量比较均匀, 适合大规模批量生产制造加氢精制催化剂。
混捏法又称干混捏法, 是将载体粉料(氧化铝干胶粉)、加氢活性组分前驱体与一定量的助剂、扩孔剂、助挤剂(田箐粉)和粘结剂(硝酸溶液)充分混合, 在挤条机上挤出成型, 再干燥焙烧制成催化剂。与浸渍法相比, 混捏法优点在于催化剂的制备只需要经过一次混捏成型、干燥和焙烧, 制备步骤大大简化, 省时省力。缺点在于部分活性组分包埋在催化剂颗粒内部, 降低了活性组分的利用率。浸渍法可以增强助催化剂与主催化剂的协同作用, 提高活性组分的分散度, 但是混捏法却无法达到活性组分的高利用率以及使助催化剂与主催化剂的协同效应最大化。综合看来, 浸渍法优势更明显, 目前加氢精制催化剂的制备方法以浸渍法为主。但是研究者对混捏法工艺又进行了改进, 形成湿混捏法。湿混捏法是先将加氢活性组分前驱体与一定量的稳定剂和粘结剂调制成糊状物, 再与载体粉料、助剂、扩孔剂和助挤剂混合, 经充分混捏后在挤条机上成型, 之后经过干燥焙烧制成催化剂。改进后的湿混捏法提高了催化剂中活性组分的利用率, 制备方法和步骤依然简化、不耗时。目前少部分加氢精制催化剂采用混捏法制备。
在加氢精制催化剂方面, 人们对催化剂加氢脱硫和加氢脱氮的活性要求更高, 需要开发更好的加氢催化剂。以加氢脱硫催化剂为例, 由于石油储量下降, 石油重质化和劣质化问题愈来愈突出, 世界范围高硫原油逐年增多, 且各国环境立法关于限制燃油中硫含量的要求日趋严格, 因此开发性能优良的超深度加氢脱硫催化剂, 不仅成为加氢脱硫领域的核心, 也使加氢处理技术在石油加工领域日益受到重视。除了火电厂和工厂排放的SOx以外, 油品中的硫化物又是空气污染的主要源头之一。燃油中的有机含硫化合物经燃烧后产生的SOx不仅能导致酸雨形成, 还能使汽车发动机尾气净化系统的三效催化剂产生不可逆中毒, 也会参与形成粉尘颗粒物PM2.5, 导致日渐增多的雾霾天气, 严重危害环境和人体健康, 因而引起广泛关注。为此, 各国都颁布了严格的燃油含硫量标准, 欧洲已于2005年实现柴油硫含量小于10× 10-6标准, 我国已于2012年6月1日在北京率先执行硫含量低于10× 10-6的京Ⅴ 清洁柴油指标, 并于2015年在全国范围全面实行相当于欧Ⅳ [< 50× 10-6]排放标准的柴油硫指标, 预计2018年在全国推广使用相当于欧Ⅴ [< 10× 10-6]排放标准的清洁柴油硫指标。
目前, 工业上一般常用的加氢脱硫催化剂有:Co-Mo/Al2O3、Ni-Mo-P/Al2O3、Ni-W-B/Al2O3、Ni-Co-Mo/Al2O3和Co-W/Al2O3等。但随着对硫含量限定的标准越来越高, 这些催化剂活性已经不能满足超深度脱硫需要, 迫切需要提高催化剂的超深度脱硫活性。
调整工艺操作条件和使用新型反应器都需要巨额的投资费用, 相比之下, 研制一种能够在现有的生产装置上按照现行的操作条件进行超深度加氢脱硫的新型催化剂, 是一种更为经济、更加可行的方法。与传统加氢脱硫催化剂相比, 多金属体相催化剂(如Nebula)显示出超高加氢脱硫活性, 能够对渣油等重油原料进行加氢处理。体相催化剂目前仍处于发展阶段, 进入工业应用的主要是Nebula体相催化剂。在过去的十几年, Nebula催化剂的工业应用增加迅速。体相催化剂与传统催化剂概念不同, 在加氢领域具有跳跃性重大发展, 开发新型体相催化剂已引起人们越来越多的关注。
4.5 催化重整中的催化作用
催化重整是以石脑油为原料, 有H2和催化剂存在时, 在一定的温度和压力条件下, 使烃类分子发生重排, 将石脑油转化为富含芳烃的重整生成油的工业过程。根据催化重整产品的特点, 催化重整过程的主要目的有:生产高辛烷值汽油组分; 生产苯、甲苯、二甲苯等单体芳烃, 为化纤、橡胶、塑料和精细化工提供原料; 生产化工所需的溶剂, 加氢所需的高纯廉价H2和液化气等副产品[1, 2]。
催化重整由于其特殊的产品结构和性能, 在炼油工业中具有重要地位。重整汽油是车用汽油的主要调和组分。北美和欧洲汽油主要由催化裂化汽油和催化重整汽油构成, 而我国催化裂化汽油则占据主导。催化重整汽油具有辛烷值高、烯烃和硫含量低的特点, 符合清洁汽油的标准要求。所以催化重整汽油是理想的汽油调和组分, 催化重整过程对于炼油厂生产清洁汽油至关重要。另外催化重整在石油化工中也具有重要地位和作用, 是生产苯、甲苯和二甲苯等一级基本有机化工原料的主要手段[6]。
根据现有的催化重整工艺和技术要求, 可作为催化重整过程的原料有直馏石脑油、加氢裂化石脑油、焦化石脑油、催化裂化石脑油和裂解乙烯石脑油抽余油。其中直馏石脑油作为重整原料具有烯烃和杂质含量小的优点, 是最理想的重整原料, 但目前较为紧缺。而加氢裂化石脑油同样具有烯烃和杂质含量小的优点, 使用其作为重整原料可以有效解决直馏石脑油的供给不足。而催化裂化石脑油具有烯烃、环烷烃和杂质含量较高的特点, 作为重整原料不是很理想, 一般经过预处理后作为催化重整的原料。
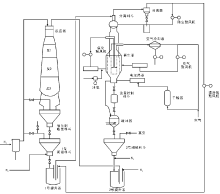
催化重整工艺根据目的产品不同主要分为高辛烷值汽油生产工艺和芳烃生产工艺, 高辛烷值汽油生产工艺由原料预处理、重整反应和重整产物分离三部分构成, 而芳烃生产工艺又在此基础上多了芳烃抽提和芳烃精馏两个部分。使用铂铼重整装置的高辛烷值汽油生产工艺流程如图4-9所示[50]。
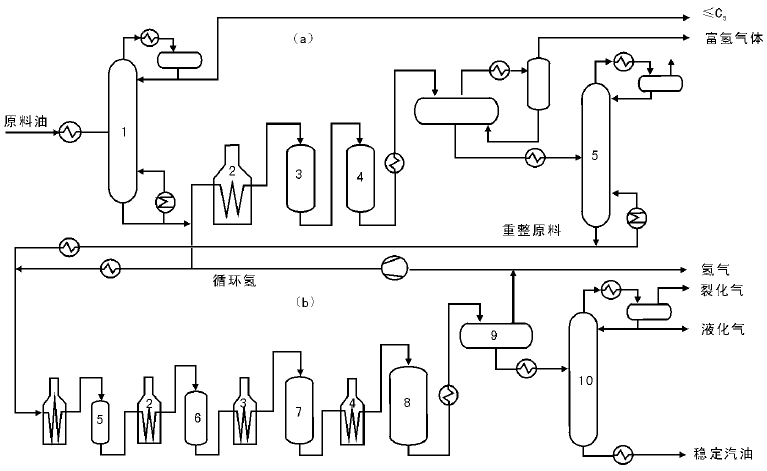 | 图 4-9 铂铼重整装置工艺流程 (a)原料预处理部分:1.预分馏塔; 2.预加氢加热炉; 3, 4.预加氢反应器; 5.脱水塔(b)反应及分馏部分:1, 2, 3, 4.加热炉; 5, 6, 7, 8.重整反应器; 9.高压分离器; 10.稳定塔 |
催化重整的发展历史主要涉及催化重整催化剂和催化重整工艺两方面, 以催化剂为主, 两者协同发展。我国催化重整催化剂的发展从固定床半再生重整使用的单铂及多金属催化剂到移动床连续重整使用的Pt-Sn/Al2O3系列催化剂都紧跟国际潮流。我国在20世纪80年代后, 用于固定床的半再生重整装置的高铼铂比工业催化剂稳定性、抗积炭能力、芳烃产率和液体收率都能达到或超过国外同类催化剂。而我国从1986年自主开发的连续重整催化剂已于1990年成功工业应用, 目前也已经开发出了一系列高稳定性高活性的连续重整催化剂[51, 52]。
4.5.1 催化重整中的化学反应
催化重整生产高辛烷值汽油或芳烃, 必须通过化学反应过程实现。在催化重整过程中发生的化学反应主要有以下5类[53]:
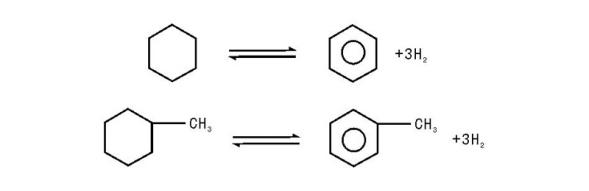
(1) 六元环烷烃的脱氢反应:
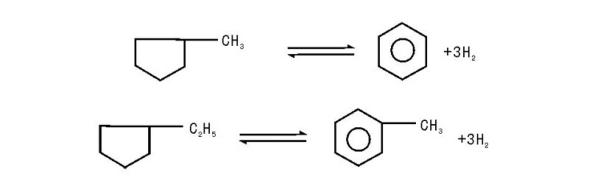
(2) 五元环烷烃的异构脱氢反应:
(3) 烷烃的环化脱氢反应:
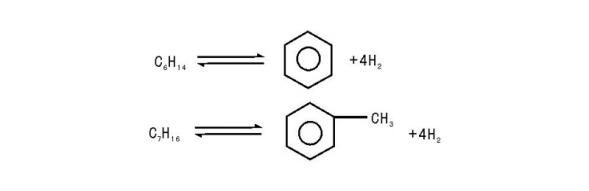
(4) 异构化反应:
n-C7H16→ i-C7H16
(5) 加氢裂化反应:
n-C8H18+H2→ 2i-C4H10
除此之外, 还会发生缩合成焦反应, 生成焦炭。
不同类型重整反应的热力学和动力学特点有所不同[54]:
(1) 六元环脱氢芳构化反应在热力学上表现为强吸热反应, 平衡常数大(800 K), 且随着温度的提高和氢油比的下降而增大, 但受压力的影响不大, 压力增大对反应不利。动力学上六元环反应速率很快, 且随着六元环碳原子数增加, 反应速率增大, 在一般的重整条件下都能达到平衡, 因此此类反应一般不受动力学控制。
(2) 五元环烷烃在重整原料中占有很大比例, 因此五元环烷烃异构脱氢芳构化是重整过程中的重要反应。五元环烷烃脱氢异构与六元环烷烃脱氢反应的热力学动力学影响因素规律相似, 反应分为异构成六元环和脱氢两部分。与六元环烷烃相比, 五元环烷烃比较容易发生加氢裂化反应, 使得芳烃的转化率降低。
(3) 理论上说碳原子数不少于6的烷烃都可以转化为芳烃, 但在非贵金属和单铂催化剂条件下其反应速率太慢, 对生产芳烃贡献较小。如今随着催化剂和重整工艺的改进, 烷烃环化脱氢生成芳烃的产量已大幅提高。烷烃脱氢环化反应的平衡常数大(800 K), 为强吸热反应, 热力学规律和环烷烃脱氢反应的规律类似。实际生产中烷烃的转化率则比平衡低很多, 一方面是由于该反应历程很长, 导致反应速率慢, 另一方面因为烷烃在重整条件下存在多个方向的竞争反应。
(4) 烷烃和环烷烃的异构化反应都是可逆的轻度放热反应, 平衡常数和速率都较小。提高反应温度, 平衡常数下降, 但异构产率会增加, 说明异构反应受动力学控制。但温度过高又使得加氢裂化反应加剧, 异构产率下降。反应压力和氢油比对异构化反应影响不大。
(5) 加氢裂化反应是中等程度的放热反应, 此类反应不可逆, 一般不考虑化学平衡, 只考虑反应速率。高温高压有利于加氢裂化反应的进行。
综上所述, 各种重整反应的顺序为:六元环烷烃脱氢> 烷烃、环烷烃异构化> 烷烃加氢裂化> 烷烃环化脱氢。
4.5.2 催化重整催化剂
4.5.2.1 重整催化剂的功能、组成和分类
工业重整催化剂根据组成可分为贵金属催化剂和非贵金属催化剂两大类, 其中, 贵金属催化剂主要有Pt-Re/Al2O3、Pt-Sn/Al2O3和Pt-Ir/Al2O3等系列; 而非贵金属催化剂主要有Cr2O3/Al2O3和MoO3/Al2O3等, 其性能较贵金属催化剂低得多[1]。早期的重整催化剂曾使用过以钼、铬为主要活性组分的催化剂, 由于活性及稳定性差, 后来逐渐停止使用。之后以贵金属铂为主要活性组分的重整催化剂在工业上被广泛使用, 催化剂的活性比钼、铬催化剂的活性高上百倍。目前, 工业上广泛使用的是以贵金属铂为基本活性组分的双金属和多金属催化剂。
贵金属催化剂由活性组分(主催化剂, 如铂)、助催化剂(如铼, 锡等)和载体(如含卤素的γ -Al2O3)构成。由于重整过程有芳构化和异构化两种类型的反应, 因此要求重整催化剂具有脱氢和裂化异构化双功能。一般由金属元素提供脱氢反应功能, 即金属功能; 由卤素提供异构化反应功能, 即酸性功能。重整催化剂的这两种功能在反应过程中要保持一定平衡, 否则会影响催化剂的整体选择性。助催化剂本身没有催化活性或活性很弱, 但与活性组分共存时能改善催化剂的活性、稳定性和选择性。另外, 引入其他金属作为助催化剂还可以减少铂含量, 降低成本。目前多金属重整催化剂主要有以下3大系列:(1) Pt-Re系列, 活性、稳定性大大提高; (2) Pt-Ir系列, 脱氢环化能力大大提高; (3) Pt-Sn系列, 低压稳定性和环化选择性好。载体一般本身不具有催化活性, 但具有较大的比表面积和较好的机械强度。目前作为重整催化剂常用的载体有η - Al2O3和γ - Al2O3。η - Al2O3比表面积大, 氯保持能力强, 但热稳定性和抗水能力差, 所以工业上重整催化剂主要使用γ - Al2O3作为载体[2]。
一般来说, 催化剂的脱氢活性、稳定性和抗毒能力随铂含量的增加而增强。由于铂是贵金属, 所以铂催化剂的制造成本主要取决于铂含量。当铂含量接近1%时, 再增加铂含量则几乎没有积极影响。随着载体和催化剂制备技术的改进, 使得活性金属组分能够更均匀的分散在载体上, 重整催化剂的铂含量也更趋于降低。目前工业用重整催化剂的铂含量大多是0.2%~0.3%。此外, 铂铼双金属重整催化剂已取代了单铂催化剂。铼的主要作用是提高催化剂的容炭能力和稳定性, 延长了运转周期, 使反应条件能够更加苛刻, 特别适用于固定床反应器。工业用铂铼催化剂中铼铂含量比一般为1~2, 较高的铼含量对提高催化剂的稳定性有利。而铂锡重整催化剂在高温、低压下具有良好的选择性和再生性能, 并且锡价格比铼便宜, 新鲜催化剂和再生催化剂不必预硫化, 生产操作简便。虽然铂锡催化剂的稳定性不如铂铼催化剂, 但也能满足连续重整工艺的要求, 因此近年来被广泛应用于连续重整装置。
改变卤素的含量可以调节催化剂的酸性功能。随着卤素含量的增加, 催化剂对异构化和加氢裂化等酸性反应的催化活性也增强。卤素的使用上通常有氟氯型和全氯型两种。氟在催化剂上比较稳定, 操作时不易被水带走, 因此氟氯型催化剂的酸性功能受重整原料含水量的影响较小, 一般氟氯型催化剂含氟和氯约1%。但是氟的加氢裂化性能较强, 使催化剂的性能变差, 因此近年来多采用全氯型催化剂。氯在催化剂上不稳定, 容易被水带走, 但是可以在工艺操作中根据系统中的水-氯平衡状况注氯以及在催化剂再生后进行氯化等措施来维持催化剂上氯的适宜含量。一般新鲜的全氯型催化剂含氯0.6%~1.5%, 实际操作中要求含氯量稳定在0.4%~1.0%。卤素含量太低时, 由于酸性功能不足, 芳烃转化率或产品的辛烷值低。虽然提高反应温度可以抵消这部分影响, 但会使催化剂的寿命显著缩短。另一方面, 卤素含量太高时, 加氢裂化反应增强, 导致液体产物收率下降。
一般来说, 载体本身并没有催化活性, 但具有较大的比表面积和较好的机械强度, 能够使活性组分很好地分散在表面上, 从而有效发挥其作用, 节省活性组分的用量, 同时也提高了催化剂的稳定性和机械强度。目前重整催化剂主要采用γ -Al2O3作为载体。载体的孔结构也很重要, 孔径过小不利于原料和产物的扩散, 而且容易在孔口处结焦, 导致内表面不能充分利用而使活性迅速下降。重整催化剂的堆积密度多为(0.6~0.8) g· cm-3。
在现代重整工业装置中, 单铂催化剂已经被淘汰, 目前使用的主要是用于固定床重整装置的铂铼催化剂和用于移动床连续重整装置的铂锡催化剂。从使用性能比较, 铂铼催化剂具有更好的稳定性, 而铂锡催化剂则具有更好的选择性和再生性能。实际中对于催化剂的选择应当综合重视其综合性能, 一般来说, 可以从以下3个方面综合评价:(1) 反应性能。对固定床重整装置, 重要的是具有优良的稳定性, 同时也要有良好的活性和选择性。催化剂的稳定性可以从容炭能力和生焦速率之比进行比较。如果使用稳定性好的催化剂, 在必要时还可以适当降低反应压力和氢油比, 从而提高液体产品收率并降低能耗。对于连续重整装置, 则要求催化剂具有良好的活性、选择性和再生性能; (2) 再生性能。良好的再生性能无论对固定床重整装置还是连续重整装置均很重要, 尤其是后者。连续重整催化剂通常3~7天就要循环再生一次, 其再生性能主要取决于热稳定性; (3) 其他理化性质。比表面积对催化剂保持氯的能力有影响; 机械强度、外形和颗粒均匀度对反应器床层压降有重要影响, 此性能对于连续重整装置尤为重要; 催化剂的杂质含量及孔结构也会影响稳定性。随着催化剂性能的不断改进, 催化重整工艺技术也有很大进步, 重整装置的效率和经济效益得到提高。表4-2列出催化重整主要工艺参数、反应器型式与催化剂发展的相互关系。反应压力和氢油比的不断降低不仅提高了重整汽油的辛烷值和收率, 还降低了装置的能耗, 提高了经济效益。
| 表 4-2 催化重整主要工艺参数与催化剂的关系 |
4.5.2.2 重整催化剂的失活和再生
在运转过程中, 催化剂的活性和选择性会逐渐变坏, 主要原因是积炭、卤素流失、中毒和老化, 其中积炭是催化剂活性下降的主要原因, 因此在运转过程中必须严格控制以降低催化剂的失活速率。催化剂的失活控制方法主要有:(1) 引入其他金属提高稳定性, 提高氢油比, 抑制积炭生成; (2) 控制好温度和催化剂氯含量, 抑制金属聚集; (3) 严格控制原料中的氧、氮、硫、砷和其他金属含量, 防止催化剂中毒。催化剂经长期运转以后, 因积炭而失活, 经烧炭、氯化等再生过程可以完全恢复活性; 但因金属中毒或高温烧结而严重失活则不能再生恢复活性。
根据红外光谱和XRD分析结果, 在重整催化剂上的积炭主要是缩合芳烃, 具有类石墨结构。积炭的主要成分是碳和氢, 氢碳比一般为0.5~0.8。催化剂的金属活性中心和酸性活性中心上都有积炭, 但积炭大部分是在酸性载体γ -Al2O3上。金属活性中心上的积炭在氢的作用下可能解聚而消除, 但是在酸性活性中心上的积炭在氢的作用下则难以除去。对于一般的铂催化剂, 当积炭增至3%~10%时, 活性大半丧失; 而对于铂铼催化剂, 积炭约达到20%时, 活性才大半丧失。催化剂因积炭引起的活性降低可以采用提高反应温度的办法补偿。但是提高反应温度有一定的限制, 一般重整装置限制反应温度不超过520 ℃。当反应温度已经提升到限制温度而催化剂的活性仍不能满足要求时, 就需要通过再生的方法烧去积炭恢复催化剂活性。再生性能好的催化剂再生后活性基本可以恢复到原有水平。催化剂上积炭的速率与原料的性质和操作条件有关。原料的终馏点高、不饱和烃含量高时积炭速率快; 反应条件苛刻也会使积炭速率加快。
催化剂的脱氢功能和酸性功能应当有良好的配合。氯是催化剂酸性功能的主要来源, 在生产过程中应当使其含量维持在适宜的范围, 氯含量过低时, 催化剂的活性下降; 氯含量过高, 加氢裂化反应加剧, 液体产物收率下降。在生产过程中, 催化剂上的氯含量会发生变化。当原料氯含量过高时, 氯会在催化剂上沉积而使催化剂氯含量增加。当原料中水含量过高或反应生成水过多时, 这些水分会冲洗氯使催化剂氯含量减小。高温条件下, 水的存在还会促使铂晶粒的长大并破坏氧化铝载体的微孔结构, 从而降低催化剂的活性和稳定性。此外, 水还会和氯生成HCl腐蚀设备, 对环化脱氢反应也有阻碍作用。为了严格控制系统中水和氯的含量, 国内重整装置限制原料油的氯含量和水含量不得超过5 μ g· g-1。现代重整装置还通过不同的途径判断催化剂上的氯含量, 然后采取注氯、注水等办法保证最适宜的催化剂氯含量, 即水氯平衡方法。工业装置上的注氯通常采用二氯乙烷、三氯乙烷和四氯化碳等氯化物; 注水通常采用醇类以避免对设备的腐蚀。
催化剂的中毒可以分为永久性中毒和非永久性中毒。永久性中毒的催化剂活性不可再恢复; 而非永久性中毒的催化剂在更换无毒原料后, 毒物可以逐渐排除而恢复活性。对于含铂催化剂, 砷、铅、铜、铁、镍和汞等金属毒物为永久性毒物, 而硫、氮和氧等非金属毒物为非永久性毒物。
在永久性毒物中砷是最应当注意的, 它与铂有很强的亲和力, 可与铂形成合金造成催化剂永久性中毒。当催化剂上的砷含量超过200 μ g· g-1时, 催化剂活性就完全丧失。铂催化剂的实验结果表明, 若要求催化剂活性保持在原活性的80%以上, 砷含量应低于100 μ g· g-1。实际上工业装置中常限制重整原料油的砷含量不超过1 μ g· kg-1。在一般的石油馏分中, 其砷含量随着沸点的升高而增加, 而原油中的砷约90%是集中在蒸馏残油中。原油中的砷化物受热会分解, 因此二次加工的汽油常含有较多的砷。铅与铂也可以形成稳定的化合物, 造成催化剂中毒。石油馏分中铅含量很少, 铅的来源主要是原料油被污染所致, 而铜、铁和汞等毒物的主要来源是管线系统内的杂质。此外钠也是铂催化剂的毒物, 所以禁止使用NaOH处理重整原料。
原料中的含硫化合物在重整反应条件下生成H2S, 若不从系统中除去, H2S在循环氢中积聚, 导致催化剂的脱氢活性下降。研究表明, 当原料中硫含量为0.01%和0.03%时, 铂催化剂的脱氢活性分别降低50%和80%。原料中允许的硫含量与系统的氢分压有关, 当氢分压较高时, 允许的硫含量可以较高。一般情况下, 硫对铂催化剂毒化是暂时性的, 一旦进料中不再含硫, 一段时间后催化剂的活性就可以恢复。但是如果长时间存在过量的硫, 也会造成永久性中毒。多数双金属催化剂比铂催化剂对硫更加敏感, 因此其对硫的限制也更加严格。硫可以与铼发生反应, 并难以用氢气还原。另一方面, 原料中的硫含量也不是越低越好, 有限的硫含量可以抑制氢解反应和深度脱氢反应, 这对铂铼催化剂非常重要。在使用新鲜的或刚再生过的铂铼催化剂之前, 常常要有控制地对催化剂进行硫化。原料中的含氮化合物在重整反应条件下转化为氨, 吸附在酸性中心上抑制催化剂的加氢裂化、异构化和环化脱氢性能。一般认为, 氮对催化剂的毒化作用是暂时的。CO能与铂形成配合物, 造成铂催化剂中毒。
在正常运转过程中, 重整催化剂表面上的积炭增多和铂晶粒的聚集会导致催化剂活性下降, 因此当催化剂的活性降低至一定程度后, 必须进行再生以恢复其活性。半再生式固定床重整装置的催化剂一般是0.5~2年再生一次, 移动床连续重整装置的催化剂一般是3~7天再生一次。虽然两者反应器的型式不同, 但再生的原理和方法相同。重整催化剂的再生包括烧焦、氯化和干燥3个工序。一般来说, 再生后的重整催化剂活性基本上可以完全恢复。
重整催化剂上焦炭的主要成分是碳和氢, 烧焦时焦炭中氢的燃烧速率远大于碳的燃烧速率, 所以烧焦时主要考虑碳的燃烧。在相同的烧焦温度和氧分压条件下, 重整催化剂上的焦炭燃烧速率比催化裂化催化剂上的焦炭燃烧速率快。在重整催化剂的再生过程中, 最重要的问题是通过控制烧焦反应速率控制反应温度, 过高的温度会使催化剂的金属铂晶粒发生聚集, 还会不可逆地破坏载体的结构。一般再生时反应器的温度不超过550 ℃。另外烧焦时还应控制循环气中O2、H2O和CO2含量。在烧焦过程中, 催化剂上的氯会大量损失, 铂晶粒也会发生聚集, 而氯化更新工序可以补充氯并使铂晶粒重新分散, 以恢复催化剂的活性。工业上一般采用二氯乙烷进行氯化, 在循环气中的浓度不高于1%。循环气采用空气或高含氧量的惰性气体。为了使氯不流失, 应控制循环气中的水含量不大于0.1%。氯化一般在510 ℃、常压进行2 h。经氯化后的催化剂还需在540 ℃、空气流中氧化更新, 使铂晶粒的分散度达到要求, 时间一般为2 h。重整催化剂再生的干燥工序一般在540 ℃进行。干燥时若循环气中含有碳氢化合物则会影响铂晶粒的分散度, 相对分子量较大的碳氢化合物的影响特别显著。采用空气或高含氧量的气体作循环气, 可以抑制碳氢化合物的影响。研究结果表明, 在氮气流下, 铂铼和铂锡催化剂在480 ℃就开始出现铂晶粒的聚集现象, 但是当氮气流中含有10%以上的氧气时, 就能显著地抑制铂晶粒的聚集, 因此催化剂干燥时的循环气体宜采用空气。
新鲜催化剂和再生后催化剂中的金属组分都是处于氧化状态, 使用前必须先还原成金属状态。还原过程约在480 ℃氢气气氛进行, 由于水的生成, 还应控制系统的含水量。铂铼催化剂和某些多金属催化剂在刚开始进油时, 可能会表现出强烈的氢解性能和深度脱氢活性, 这会导致催化剂床层温度的剧烈上升以及催化剂的迅速积炭, 导致活性和选择性变差。因此在进油之前必须对催化剂进行预硫化以抑制其氢解性能和深度脱氢活性。而铂锡催化剂不需要预硫化, 因为锡可以起到与硫相当的抑制作用。预硫化采用硫醇或二硫化碳作为硫化剂, 用预加氢精制油稀释后加热送入反应系统。硫化剂使用量一般为10-6级别, 预硫化温度(350~390) ℃, 压力(0.4~0.8) MPa。
4.5.3 催化重整原料的选择和预处理
由于催化重整生产方案和重整催化剂的不同以及重整催化剂昂贵易失活, 为了提高装置的运转周期和产品收率, 必须对重整原料进行选择和预精制处理。
重整原料的选择主要分为3个方面:馏分组成、族组成和毒物杂质含量。重整原料馏分组成的要求由生产目的确定, 以高辛烷值汽油为目的时, 一般以直馏汽油为原料, 馏分范围(90~180) ℃; 以芳烃为目的时, 馏分范围(60~145)℃。在重整过程中, 芳构化速率有差异, 其中环烷烃的芳构化速率快, 对芳烃收率贡献大, 而烷烃则相反。一般以芳烃潜含量表示重整原料的族组成, 芳烃潜含量越高, 重整原料的族组成越理想。
重整原料中含有少量的氮、硫和砷等杂质会使催化剂失活, 因此必须对原料进行除杂预处理。重整原料的预处理由预脱砷、预分馏、预加氢和脱水等单元组成。砷是重整催化剂和各种预加氢精制催化剂最致命的毒物, 因此重整反应的原料必须严格控制砷含量(10-9以下)。目前工业上主要使用的预脱砷方法主要有:(1) 吸附法, 使用浸渍有CuSO4的硅酸铝小球作为脱砷吸附剂; (2) 氧化法, 将氧化剂与原料混合, 再经过蒸馏或水洗将砷的氧化物除去; (3) 加氢法, 将加氢预脱砷反应器和加氢预精制反应器串联, 使用脱砷剂将砷吸附。
4.5.4 催化重整工艺流程
工业重整装置根据反应器类型可分为固定床和移动床反应工艺过程。根据催化剂再生方式又可分为半再生、循环再生和连续再生工艺过程[55]。
固定床半再生式重整工艺流程采用轴向或径向固定床反应器, 使用条形或球形催化剂。当催化剂失活时必须停运装置, 对催化剂进行再生处理。这种工艺具有系统简单、运转操作维护方便、建筑费用低等优点, 但由于催化剂的失活限制其长期连续运转。固定床半再生式重整工艺以麦格纳重整工艺流程为例(图4-10)。将循环氢分为两路, 分别从第一和第三反应器进入, 第一、第二反应器采用高空速、低温度和低氢油比, 有利于环烷烃的脱氢反应, 同时抑制了加氢裂化反应。后面两个反应器采用低空速、高温度和高氢油比, 有利于烷烃的脱氢环化反应。这种装置的主要特点是高液体收率、装置能耗降低。
半再生式重整会因催化剂失活而被迫停工, 因此为了保持催化剂的高活性, 自20世纪70年代分别由美国环球油品公司(UOP)和法国国家石油研究院(IFP)研究并发展了移动床反应器连续再生式重整工艺。这种工艺设有催化剂再生器, 使催化剂在反应器和再生器之间不断地循环。目前世界上连续重整有重叠式(美国UOP, 图4-11)和并列式(法国IFP, 图4-12)两种工艺, 都设有单独的催化剂连续再生系统; 而在具体工艺流程和设备结构方面则有以下差异:
(1) UOP反应器为重叠式布置, 占地面积小, 催化剂靠重力流动, 设备框架比较高; IFP反应器为并列式布置, 占地面积大, 催化剂的输送靠气体的提升, 设备高度低, 维修方便。
(2) UOP再生气采用热循环, 流程简单, 但对设备材质要求高, 再生气水含量高; IFP再生气采用冷循环, 设备多, 但对材质要求低, 再生气水含量低, 有利于催化剂比表面积的保持。
(3) UOP装置的闭锁料斗设在反应器底部, 再生压力比反应压力低; IFP装置的闭锁料斗设在反应器上部, 再生压力比反应压力高, 有利于催化剂的烧焦。
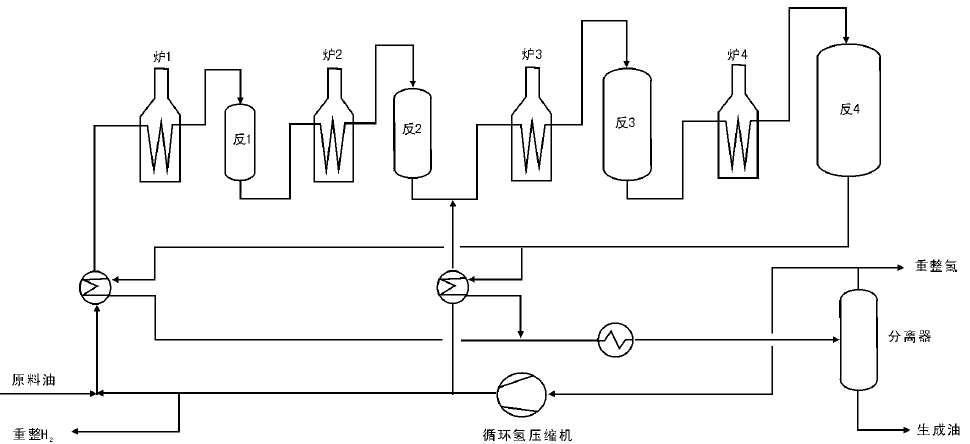 | 图 4-10 麦格纳重整反应工艺流程 |
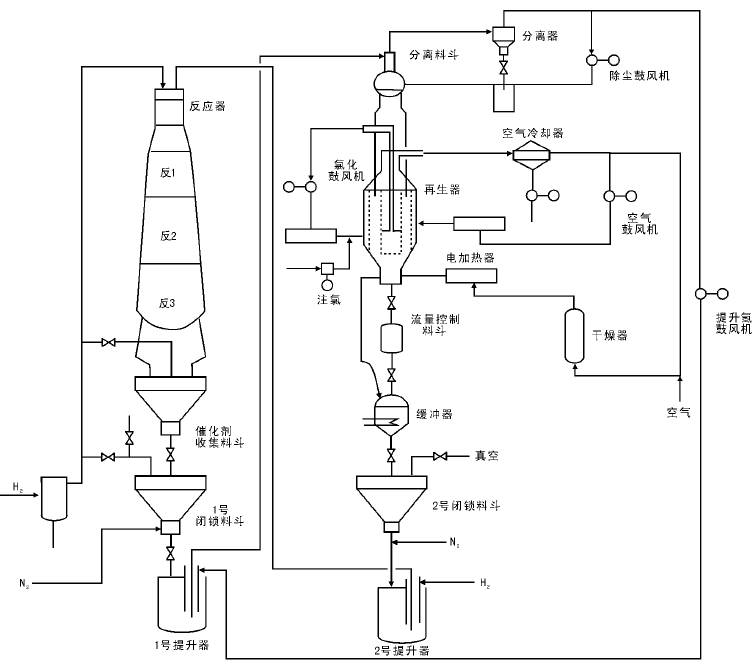 | 图 4-11 UOP连续重整反应系统流程 |
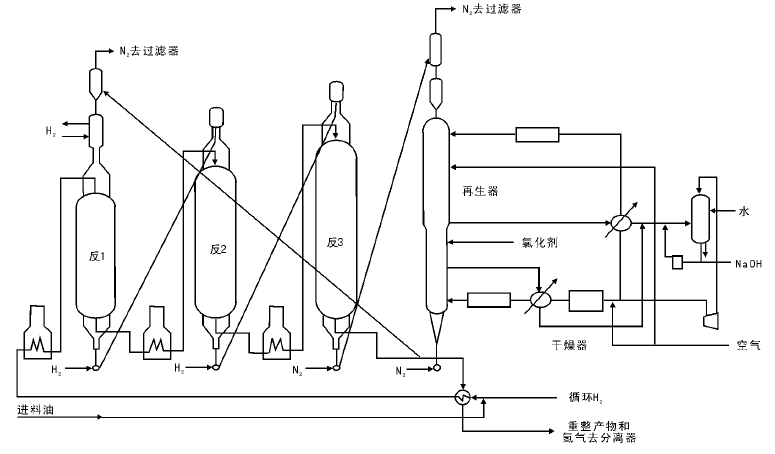 | 图 4-12 IFP连续重整反应系统流程 |
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|