{kind=link}
{kind=link}
{kind=link}
{kind=link}
第六章 合成气化学
引用本文
葛庆杰. 第六章 合成气化学[J]. 工业催化, 2016,24(3): 82-104. 
Permissions
Copyright©2016, 《工业催化》编辑部
《工业催化》编辑部 所有
第六章 合成气化学

作者简介:
葛庆杰,男,1971年生,博士,中国科学院大连化学物理研究所研究员,博士研究生导师。
长期从事多相催化的应用基础研究,以具有重要科学意义和应用背景的能源化工反应为对象,进行催化新材料、新工艺、新技术等科学研究,为能源资源的合理化应用提供新型催化材料、工艺和技术。研究方向主要为:合成气转化制洁净液体燃料和化学品,合成甲醇耐硫催化新材料,多功能催化材料的制备化学,烃类选择氧化制合成气和氢,氢气的制备与分离,烃类催化脱氢新材料等。近年来,承担多项国家、科学院和企业项目,在ACS Nano、Nanosacle、Applied Catalysis等期刊发表研究论文120篇,申请发明专利40余项,培养硕士和博士研究生10名。
6.1 前 言
随着社会技术的进步和经济的发展, 能源与环境已成为关乎人类社会能否可持续发展的两大课题。目前世界能源消费结构仍以化石能源为主, 社会发展和环境安全之间的矛盾和冲突愈演愈烈, 因此, 化石能源特别是煤的清洁利用和可再生清洁能源被提到前所未有的高度。而就化石能源而言, 特别是石油, 由于过去近百年的过度开采和无节制使用, 已呈现匮乏趋势, 迫使人们探寻由非石油路线来获得通常由石油提供的液体燃料和烯烃等重要化工产品[1, 2]。
第二次工业革命后, 人类社会使用能源先后经历了煤(固体)、石油(液体)和天然气(气体)燃料三个主要时期。在经历煤和石油两个时期后, 石油资源匮乏的趋势和供应危机日益明显, 迫使人们寻找非石油路线制取液体燃料和烯烃等重要化工产品的新途径, 天然气作为最清洁的化石燃料, 在开发替代石油路线新过程中成为首选。1985年天然气经甲醇制汽油(MTG)问世, 从此在天然气和石油之间架起了一座具有里程碑意义的桥梁。随后天然气经合成气F-T合成制液体燃料(GTL)与甲醇制烯烃(MTO)相继实现工业化, 似乎预示着天然气时代的到来。但实际发展情况表明事情要复杂得多, 首先是GTL经济性一直受到油价涨落的影响, 其次天然气本身价格常常也随油价上涨而上涨。煤作为替代石油的另一重要能源资源在世界上具有丰富的储藏量, 煤转化重新被提上日程, 尤其是对于缺油、相对少气和富煤的我国来说格外受到关注[3]。
全球气候暖化问题在20世纪末期被集中提出, 且由于涉及人类社会能否可持续发展, 而成为从政府、企业到个人都不能回避的紧迫课题。由于化石能源(石油、天然气或煤)使用终端注定要排放CO2, 只是程度有所不同而已, 因此, 寻找非化石能源或称可再生能源, 成为能源领域又一重大课题。生物质能源(如生物柴油、生物乙醇等)由于CO2净排放为零, 被认为是替代化石能源的首选。太阳能、风能、核能发电以及太阳能直接光解水制氢, 开辟完全无碳排放的氢能时代虽然遥远, 但却是人类努力的方向, 在未来几十年应当会有所突破。据有关机构预测, 到2050年化石能源仍将占世界能源结构的50%, 而可再生能源将争取达到50%[4]。
预计未来几十年将是化石能源和后化石能源(可再生能源)等多种形式能源并存和交叉的时期, 对于从事化石能源转化和有关催化技术的研究者, 今后的努力方向是化石能源的清洁制取和清洁利用, 特别是非石油路线制取液体燃料和重要化工品的研究。
6.2 合成气中枢[3]
6.2.1 合成气中枢的概念
最早公认的“ 气制液体燃料(GTL)” 是专指从天然气出发, 经由合成气F-T法合成宽馏分液态烃, 再经过进一步加工生产液体燃料和其他油品的技术。其实此前天然气经甲醇制汽油(MTG)以及更早期的天然气经合成气生产甲醇和二甲醚等都有可能成为候选液体燃料, 这是广义GTL。20世纪末甲醇制烯烃(MTO)技术的提出, 则又将GTL延伸为“ 气制化学品(GTC)” 。GTC在应对石油短缺危机方面具有里程碑意义。现在还有将煤经合成气F-T合成油称为“ CTL” 。
需要指出的是:实际上, 目前绝大多数GTL或CTL工业过程都是基于经过合成气的间接法。因此, 称“ 合成气制化学品(SGTC)” , 当比笼统的GTL或CTL说法更为确切。与已有百年历史的煤锅炉燃烧/气轮机发电相比, 合成气燃气轮机/蒸汽轮机联合发电(IGCC)显然有更高的能量转化效率和更低的污染物排放, 而受到愈来愈多的重视。这就是更广泛意义上的“ 合成气制能源(SGTE)” 。
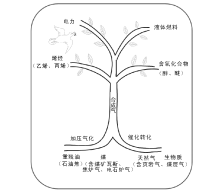
因此, 开辟多种资源如天然气、煤层气(CBM)、页岩气、煤(包括衍生的焦炉气、电石炉气和煤矿瓦斯抽放气)、重残油(包括石油焦)以及生物质等制取合成气, 再进一步合成液体燃料、重要化学品或发电, 或多联产(即SGTC, SGTE), 应是一个适合中国国情的对策。合成气中枢的新概念也相应提出(见图6-1)。
合成气中枢(Syngas Hub)是将天然气、煤和生物质等多种原料气化生产合成气, 再以合成气为原料, 选择合成需要的液体燃料和化工产品。合成气成为连接天然气、石油、煤炭、生物质等上游资源和下游产品液体燃料、乙烯、丙烯、醋酸和芳烃等化工产品的枢纽。相对于传统石油炼制、石油化工、煤化工和天然气化工等一个个单独领域的概念, 它是一个跨行业的、综合程度更高的新概念。
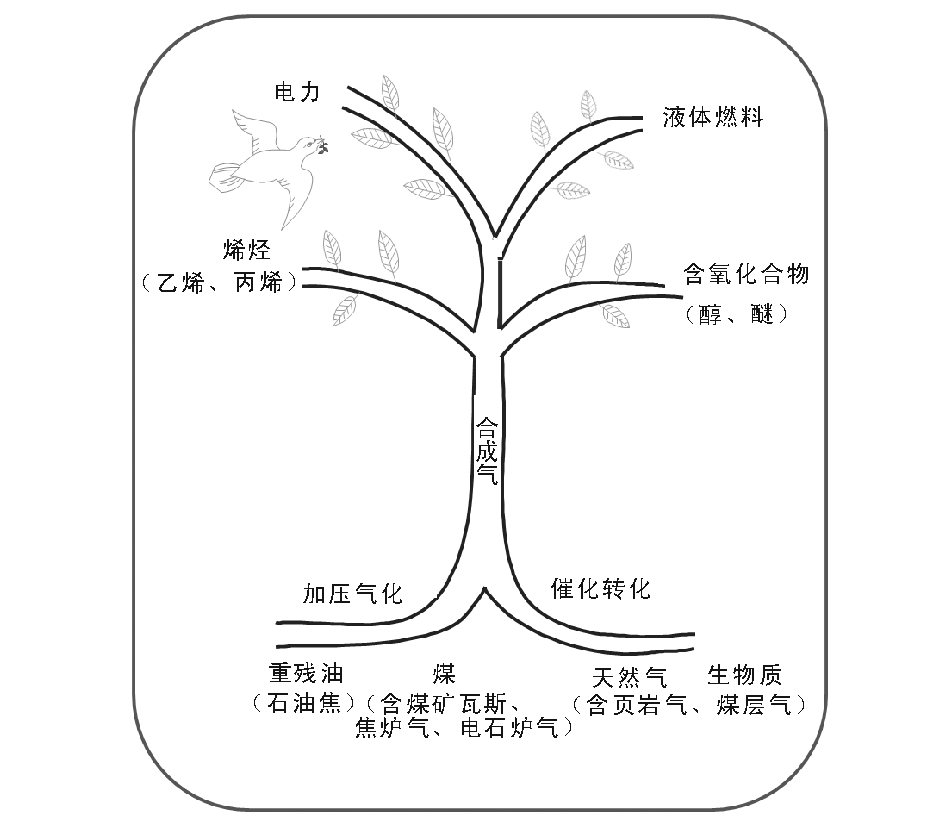 | 图6-1 合成气中枢(Syngas Hub) |
6.2.2 合成气中枢的催化技术
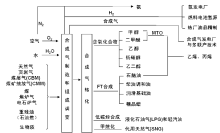
合成气中枢有两大主要任务, 即合成气制造和组成调变以及合成气转化利用。图6-2列出合成气中枢两大任务所涉及的反应过程, 这些过程绝大多数是通过相应的催化技术实现。
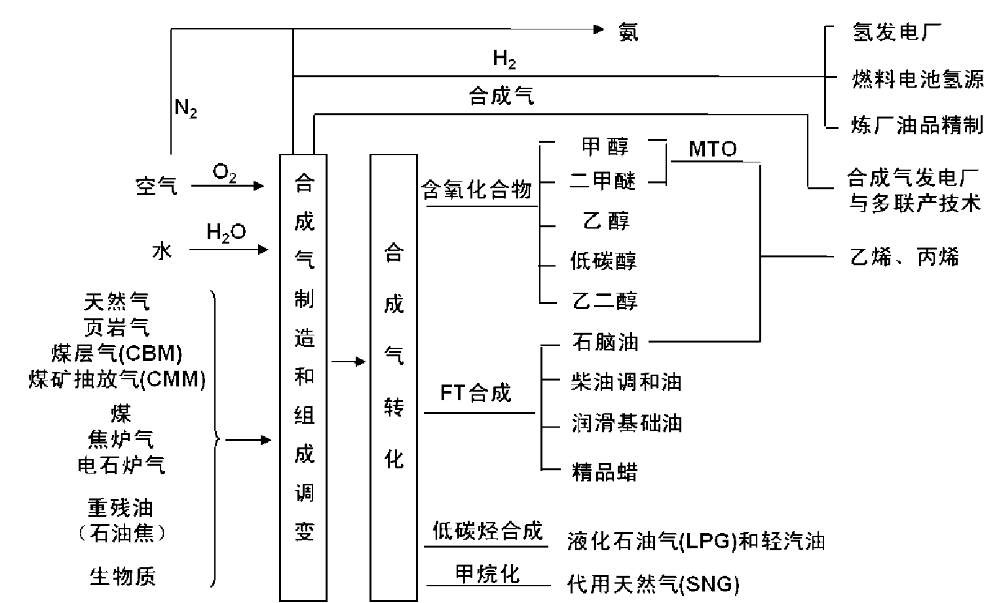 | 图6-2 合成气中枢的催化技术 |
6.3 合成气制造
合成气以CO和H2为主要组分, 原料范围较广, 既可由煤、焦炭或生物质等固体燃料产生, 又可由天然气、煤层气、页岩气和石脑油等轻质烃类制取, 还可由重油经部分氧化法生产。其生产投资和成本通常占下游产品成本的50%~60%, 廉价合成气生产技术研究极其重要[5]。
目前, 天然气(甲烷)水蒸汽重整最成熟, H2/CO比高, 一般在2.5~3.0, 但设备昂贵; 甲烷自热重整和催化部分氧化制合成气技术仍在开发中; 煤气化炉设备复杂、投资大、污染重, H2/CO比偏低, 约0.4~0.7; 生物质(碳水化合物)制合成气是一个新课题。此外, 以煤为原料生产的焦炉气、电石炉气、地下煤气化水煤气等都是含不同H2/CO比的合成气, 中枢将对各种来源的合成气选择性地进行各种下游产品所需H2/CO比例的最佳调变, 实现资源和能源的最佳利用(见表6-1)[6, 7, 8, 9, 10, 11]。
| 表6-1 不同原料来源的合成气组成 |
从表6-1中可以看出, 天然气蒸汽转化、地下煤气化两段法和焦炉气都是高H2含量合成气, 而粉煤气化、电石炉气均属高CO含量合成气。如各自进行下游合成, 为满足甲醇、F-T合成需要的H2/CO比为2的合成气, 前者需分离出一部分H2, 而后者则必须进行相当量的水气变换反应; 如将高H2和高CO进行混合, 将无需额外措施而取得合适的H2/CO比进行下游合成, 这对资源有效利用和能量转化效率自然十分有利。
6.3.1 煤气化制合成气
煤气化是指以煤或煤焦为原料, 以氧气(空气、富氧或工业纯氧)、水蒸汽作为气化剂, 在高温条件下通过化学反应将煤或煤焦中的可燃部分转化为可燃性气体(合成气, 又称煤气)的过程[12]。该过程已有150多年历史, 是20世纪50年代以前主要的合成气生产方法。
煤炭气化必须具备3个条件, 即气化炉、气化剂和供给热量, 三者缺一不可。煤气化过程发生的反应包括煤的热解、气化和燃烧反应。煤的热解是指煤从固相变为气、固、液三相产物的过程。煤的气化和燃烧反应则包括两种反应类型, 即非均相气-固反应和均相气相反应。煤气化过程可用下式表示:
煤+气化剂→ C+CH4+CO+CO2+H2O+H2
国内外先后开发了100多种煤气化方法, 根据不同分类标准, 煤气化方法有不同分类方法, 如根据气化技术可分为地面气化和地下气化, 根据气化介质可分为富氧气化、纯氧气化、加氢气化和水蒸汽气化等, 根据传热方式可分为外热式气化、内热式气化和热载体气化, 根据气化炉可分为移动床(固定床)气化、沸腾床(流化床)气化、气流床气化和熔融床气化等。其中, 煤炭地面气化技术是目前常用技术, 应用较多的有壳牌粉煤气化技术、德士古水煤浆气化技术和加压气流床气化技术; 煤炭地下气化生产合成气(包括空气煤气和水煤气)是煤炭开发利用的一个方向。
影响煤气化反应性能的因素包括煤化度、煤组成、煤热解和预处理条件、气化剂类型及含量、气化炉等。混合煤气的H2/CO体积比约为0.5, 水煤气的H2/CO体积比约为1.5。
煤气化制合成气可广泛用作工业燃气、民用煤气、化工或燃料合成原料气、冶金用还原气、联合循环发电燃气等。
6.3.2 天然气(含煤层气、页岩气等)制合成气
主要成分为甲烷的天然气(含煤层气、页岩气等)制合成气的典型过程有:甲烷水蒸汽重整制合成气 (式6-1)、甲烷CO2重整制合成气(式6-2)和甲烷部分氧化制合成气(式6-3)[13]:
CH4+H2O=CO+3H2
△
CH4+CO2=2CO+2H2
△
CH4+0.5O2=CO+2H2
△
从上述反应方程式可看出, 甲烷水蒸汽重整和甲烷-CO2重整反应均为吸热反应, 同时是分子数增大反应, 因此低压、高温有利于反应的进行; 而甲烷部分氧化制合成气反应是微放热、分子数增大反应, 低温、低压有利于反应的进行。
6.3.2.1 甲烷水蒸汽重整制合成气
目前, 甲烷水蒸汽重整过程是工业上天然气制合成气的主要途径。该方法的基本原理为:甲烷与水蒸汽在催化剂存在及高温条件下反应生成合成气。它是一强吸热过程, 通常在> 800 ℃的高温条件下进行, 为防止催化剂积炭, 一般采用高水碳比操作[V(H2O):V(CH4)=2.5~3], 所得合成气中V(H2):V(CO)≈ 3, 适合于合成氨及制氢过程, 而用于甲醇合成及F-T合成等重要工业过程不理想。甲烷水蒸汽重整制合成气的工艺过程能耗高、投资大、生产能力低[14]。
工业采用的甲烷水蒸汽重整催化剂多为负载型催化剂, 活性组分主要是Ni、Co、Fe、Cu 等非贵金属和Rh、Ru、Pt 等贵金属, 前者较后者的活性和抗积炭性能略差, 但由于其价格低廉、原料易得, 所以被广泛应用, 尤其是Ni催化剂[15]。催化剂的助剂和载体对催化剂的性能、强度、密度及耐热性能等性质均有影响。助剂可抑制催化剂的熔结过程, 防止活性组分晶粒长大, 能够增加活性中心对反应物的吸附, 进而增强甲烷的活化裂解过程和催化剂的抗积炭性能, 并延长催化剂使用寿命[16]。目前催化剂选用的助剂有Na2O、K2O 等碱金属, MgO、CaO 等碱土金属, ZrO2等稀有金属氧化物和CeO2、La2O3等稀土金属氧化物[17]。载体对催化剂活性组分不仅起物理支撑及分散作用, 而且通过载体与金属间电子效应及强相互作用(SMSI)改善催化剂的物理化学性能, 载体需要具有良好机械强度和抗烧结能力, 目前研究较多的载体有Al2O3、TiO2、ZrO2、La2O3、MgO、SiO2、CaO、ZSM-5 沸石分子筛及镁铝尖晶石等。
Hayashi H等[18]采用水-油乳液制备的Ni/Al2O3催化剂, 可在低水碳比条件下长时间保持高催化活性, 反应40 h后只有微量积炭, 催化剂增重量< 0.3%; 而采用浸渍法制备的Ni/Al2O3催化剂反应20 h后, 催化剂增重量> 30%。Parizotto N V等[19]研究了添加了Ag助剂的Ni/Al2O3催化剂在低水碳比(1:2)、温度600 ℃条件下的甲烷水蒸汽重整反应, 结果表明, 当Ag质量分数> 0.3%时, 催化剂具有较强的抗积炭性能, 6 h内甲烷转化率保持不变, 但转化率较低, Ag质量分数为0.6%时, 甲烷转化率约18%。赵云莉等[20]在固定床反应器中考察了助剂MgO、CaO对Ni/γ -Al2O3催化剂的甲烷水蒸汽重整反应性能影响, 结果表明, CaO的存在可增加催化剂活性组分NiO的还原性和分散性能, 提高Ni/γ -Al2O3催化剂的抗积炭性能。王大文[21, 22]制备了不同MgO改性的Ni基整体式催化剂, 研究了La助剂对Ni基整体式催化剂性能的影响, 结果表明, La助剂可明显促进Ni基整体式催化剂的甲烷水蒸汽重整反应性能。吴俊明等[23]研究了一种低镍质量分数的Ni-Mg-O固溶体甲烷水蒸汽反应催化剂, 在750 ℃下, 甲烷水蒸汽反应的转化频率为64 s-1, 在高温850 ℃和低水碳比1.0 条件下, 反应60 h后催化剂的活性无明显降低, 且催化剂表面无积炭产生, 这与催化剂表面存在的粒径较小的镍金属颗粒有关。Christensen K O等[24]研究了复合载体催化剂Ni/MgO-Al2O3, 发现镁铝水滑石结构的MgO-Al2O3粒径较小, 提高了Ni的分散度, 较大程度改善了传统Ni/Al2O3、Ni/MgO催化剂的反应性能。Laosiripojana N等[25]制备了甲烷水蒸汽重整反应Ni/Ce-ZrO2催化剂, 该催化剂显示出优于常规Ni/Ce-ZrO2和Ni/Al2O3催化剂的抗积炭能力, 这与高比表面积Ce-ZrO2载体具有高储氧能力有关。重整反应中, 除Ni表面发生反应外, Ce-ZrO2表面也发生晶格氧(Ox)与催化剂表面吸附甲烷之间的氧化还原反应, 该反应可有效抑制Ni表面积炭的形成。Young-Sam Oh等[26]指出, Ni/Ce-ZrO2催化剂加入θ -Al2O3可增加其机械强度, 并通过适当预处理可使催化剂Ni/Ce-ZrO2/θ -Al2O3具有较高催化活性与抗积炭性能。
Craciun R等[27]报道, CeO2对Pd/γ -Al2O3的甲烷水蒸汽重整催化性能有促进作用:Pd和CeO2在γ -Al2O3上的结构和分散状态对催化剂活性和失活有影响, 不同价态Pd的比例和分散状况依赖于CeO2的结构。Pd在晶体CeO2(粒径< 2 nm)上能很好分散, 但在无定形CeO2[(粒径为(10~20) nm]上, Pd会团聚成较大颗粒。由于Pd与CeO2的协同作用, 在Pd/CeO2/γ -Al2O3催化剂上进行甲烷水蒸汽重整的反应速率相对于在Pd/γ -Al2O3催化剂上可提高两个数量级。Berman A等[28]考察了Ru/(α -Al2O3+MnOx)催化剂甲烷水蒸汽重整反应的稳定性和动力学性质, 发现在1 100 ℃下经过100 h后保持良好的反应活性, 在催化剂表面上甲烷和水发生吸附和解离的同时伴随着吸附氧和碳。
综上所述, 甲烷水蒸汽重整制合成气研究重点在于低水碳比条件下高活性和高稳定性催化剂的研制以及为合理利用能量和降低装置设备投资而进行的工艺、反应设备等方面的改进, 反应机理和动力学模型建立等。
6.3.2.2 甲烷-CO2重整制合成气
甲烷-CO2重整制合成气是天然气重整制合成气的又一反应过程, 基本原理是甲烷与CO2在催化剂存在及高温条件下反应生成合成气。它是一强吸热过程, 通常在> 800 ℃的高温条件下进行, 所得合成气中V(H2):V(CO)≈ 1, 适合于羰基合成和F-T合成制化学品。
根据热力学数据分析, 反应(6-2)是独立的吸热反应, 高温对反应有利, 且只有高于645 ℃才是热力学上可行的反应。高温下Δ S0=257 J· (mol· K)-1, 很大的熵变意味着反应可进行彻底。然而过高的反应温度不仅会造成高能耗, 而且对反应器材质也提出更高要求, 降低反应温度减少能耗的最有效办法是选择适宜催化剂。
从文献报道看, 甲烷-CO2重整制合成气的催化剂一般采用除Os外的Ⅷ 族过渡金属作为活性组分, 其中采用金属Ni为催化剂的报道最多[29]。早在1928年, Fischer和Tropsch就将Fe、Co、Ni、Cu、Mo等负载在黏土、硅石和碳酸镁耐高温混合物上, 发现在温度(860~1 000) ℃, 以Ni和Co为活性组分、Al2O3为助剂的催化剂对甲烷-CO2重整制合成气具有较好活性[30]。随后研究表明, 非贵金属活性组分中, Ni催化剂活性最好, 寿命最长[31]。负载型贵金属催化剂则表现出更高的活性、选择性和抗积炭性能[32, 33], Solymosi F等[34]给出了贵金属对甲烷-CO2重整制合成气的催化活性顺序为:Ru> Rd> Rh> Rt> Ir。Bhat R N等[35]在负载Rh的Y分子筛催化剂上, 650 ℃条件下, 获得了90%的甲烷转化率和接近1:1的H2/CO比, 催化剂性能稳定, 无积炭生成。Tsipouriari V A等[36]研究了载体对Rh催化剂分散度的影响, 指出甲烷-CO2重整反应对金属催化剂的结构具有敏感性, Rh催化剂的特殊活性受其粒子大小影响, 同时结构敏感性受载体影响, 即载体与金属之间的相互作用直接或间接参与反应步骤。
由化学式(6-2)可知, 甲烷-CO2重整制合成气反应为吸热反应, 只有在高温下才能获得足够高的合成气收率, 而在较高的操作温度下要求催化剂载体必须具有很高的热稳定性。因此一般选择Al2O3、SiO2、MgO、CaO、TiO2、硅石、稀土氧化物以及一些复合金属氧化物和分子筛等熔点较高的物质作为催化剂载体。载体的结构与性质, 载体与金属组分的相互作用以及由此而引起的催化剂体相结构、组成、颗粒大小、分散度的变化就构成了对活性组分可还原性及其反应活性、选择性和抗积炭性等的可能影响因素。
载体作为负载型金属催化剂不可缺少的组成部分, 不仅起着支撑和分散活性金属组分的作用, 而且载体和金属组分之间互相作用对催化剂的活性和抗积炭性能均有影响。唐松柏等[37]研究表明, 载体表面的酸碱性质对催化剂的抗积炭性能有较大影响, 载体Al2O3的表面碱性强于SiO2, 两种催化剂Ni/Al2O3和Ni/SiO2在相同条件下使用后发现, 前者抗积炭性能明显强于后者。用碱性氧化物CaO和MgO对SiO2载体进行改性后发现, 催化剂的抗积炭性能得到了改善。说明在该反应中催化剂表面碱性的增强, 可提高催化剂的催化活性和抗积炭能力。
为进一步提高非贵金属催化剂活性和抗积炭能力, 在催化剂设计时, 通常加入少量催化剂助剂。常用助剂主要是碱金属和碱土金属氧化物, 多采用K2O、MgO和CaO, 还有一些稀土金属氧化物, 如CeO2、La2O3和混合稀土等。研究表明, 碱金属和碱土金属氧化物助剂的加入, 可中和催化剂的表面酸性, 降低甲烷脱氢活性, 增大CO2在催化剂表面的吸附量, 提高CO2的消炭作用。稀土金属氧化物的加入, 一方面可提高Ni的分散度, 另一方面可增加Ni2+的电子密度, 改善活性中心的缺电子状态[38]。
助剂在起到提高催化剂活性、选择性和稳定性的同时, 在甲烷-CO2重整制合成气反应中, 更多地发挥着抑制积炭、延长催化剂寿命的作用。许峥等[39]考察了碱性助剂K2O、Li2O、MgO、La2O3和CeO2对Ni/γ -Al2O3催化剂上甲烷-CO2重整反应活性和抗积炭能力的影响, 结果表明, 这些助剂对催化剂的抗积炭能力都有明显的改善作用, 其中MgO、La2O3和CeO2助剂的作用最为明显。助催化剂抑制积炭的作用既与助剂本身的酸碱性质有关, 也与助剂与活性组分的相互作用所引起的催化剂结构敏感性有关。归结起来, 两者都将导致CO2分子在催化剂表面的活性吸附, 一般认为这是助剂抑制积炭的原因。
反应过程中催化剂存在的严重积炭问题是甲烷-CO2重整反应制合成气研究的另一焦点, 催化剂的表面积炭会造成活性位的覆盖或催化剂孔隙堵塞, 进而导致催化剂活性下降乃至丧失。为此设计催化剂以及选择适宜载体、助剂时, 必须一方面能够降低甲烷脱氢积炭活性; 另一方面提高CO2吸附消炭活性, 以达到最终抑制积炭的目的。催化剂的积炭过程是一个非常复杂的物理化学过程, 引起和影响催化剂积炭的因素很多, 其中催化剂本身的组成、结构和性质是影响抗积炭的首要因素。大量研究结果表明, 负载型非贵金属催化剂易于积炭, 因而活性下降很快, 贵金属催化剂则有很强的抗积炭性能[38]。Aschcroft A T和Vernon P D F等分别研究了负载于Al2O3上的Ni、Pd、Ru、Rh和Ir催化剂在甲烷与CO2反应中的积炭行为, 结果发现Ni和Pd催化剂因积炭而很快失活, 而贵金属Ru、Rh和Ir催化剂都有很高的抗积炭性能, 保持了高活性[40, 41]。Rostroupnielsen J R等[42]研究负载于MgO上的Ni、Ru、Rh、Pd、Ir和Pt催化剂在甲烷-CO2重整反应中的积炭行为表明, 在反应条件下, Ni催化剂积炭速率最快, Pd催化剂次之, Ir和Pt等则相当缓慢。贵金属催化剂具有很强的抗积炭性能, 这是由于碳在贵金属催化剂上的溶解性特别小的缘故[43, 44], 也有研究认为, 催化剂失活有许多相互关联的因素, 积炭的速率和形式则是结构敏感性问题[45]。
总之, 甲烷-CO2重整制合成气反应的特点可概括为:(1) 强吸热反应, 只有温度大于645 ℃热力学上才可行; (2) 过高反应温度不仅会造成高能耗, 而且对反应器材质也提出了更高的要求; (3) 降低反应温度、减少能耗的最有效办法是选择适宜催化剂。目前该过程的工艺改进主要有两个方向, 一是将CH4、H2O和CO2混合转化, 水蒸汽的存在可减小积炭的危害, 还可在一定范围调节合成气中的氢碳比; 二是利用甲烷部分氧化反应的放热与甲烷-CO2重整反应的吸热耦合, 使甲烷、CO2和O2的转化反应在自热条件下进行, 可有效减少能耗, 降低反应温度, 提高反应空速, 从而缩小反应装置, 降低设备投资。甲烷-CO2催化重整制取合成气反应的研究取得了很大进展, 但距离工业化尚存在差距, 主要问题是催化剂容易因积炭和烧结而失活, 因此研制高活性、高选择性及抗积炭性的催化剂一直是科研工作者追求的目标。随着测试手段的不断进步, 人们对甲烷-CO2重整反应的研究不断深入, 如对活性中心、载体效应和助剂的作用等方面研究已取得了可喜成果, 对反应中存在的一些问题也逐步达成共识。但是反应机理、速控步骤以及积炭规律等一些理论问题尚存争论, 有待进行深入研究。
6.3.2.3 甲烷部分氧化制合成气
甲烷部分氧化法制合成气是一个温和放热反应, 反应可在较低温度[(750~ 800) ℃]下达到90%以上的热力学平衡转化率。该法具有能耗低, 反应速率快, 生产强度大, 催化剂用量小, 合成气氢碳比适合甲醇、F-T合成等优点。
甲烷部分氧化法制合成气的关键在于高活性、抗积炭催化剂的开发, 甲烷部分氧化制合成气催化剂主要分为两大类:(1) 贵金属催化剂。贵金属催化剂由于具有活性和选择性高、稳定性好、抗积炭能力强等优点受到关注。研究发现, 催化剂表面的抗积炭能力Rh、Ru、Pt、Ir≫Pd, 其中Rh、Pt 的活性和稳定性较好[46]。Yan Q G等[47]研究发现Rh的催化性能比Ru好, 认为贵金属催化剂之所以表现出不同的催化性能, 主要是由于催化剂表面发生的反应机理不同。他们在研究Rh/SiO2、Ru/SiO2催化剂表面发生的反应后发现, 在Rh/SiO2催化剂的表面, CO为最初产物, 而在Ru/SiO2催化剂的表面, CO2为最初产物, 即Rh/SiO2催化剂表面发生的是直接氧化反应, 而Ru/SiO2催化剂表面发生的是间接氧化反应。由于贵金属催化剂负载量高, 价格昂贵, 因此难以工业化应用; (2) 非贵金属催化剂。非贵金属催化剂主要是Ni、Co、Fe, 活性依次为Ni> Co> Fe。Ni 基催化剂的催化活性最佳, 仅次于贵金属铑, 但镍基催化剂存在易积炭、活性组分易流失等缺点[48]。因此, 如何提高Ni基催化剂的稳定性成为甲烷部分氧化反应研究的一个热点, 也是该催化剂能否实现工业化的关键。Choudhary V R等[49]在锆复合氧化物上负载几种金属氧化物(NiCoMgCeOx)用于甲烷部分氧化制合成气反应, 取得活性与稳定性俱佳的效果。Guo C L等[50]采用柠檬酸络合法制备钙钛矿型La2NiO4催化剂, 具有良好的抗积炭性能。尚丽霞等[51]报道了添加碱土金属助剂可减小Ni晶粒粒径, 使催化剂吸附CO2的能力有所降低, 又对催化剂上积炭起到了一定的抑制作用。严前古等[52]对热稳定性好、导热性好的惰性材料进行了研究, 如MgAl2O4等。季亚英等[53]考察了不同反应条件对Mg调变的Ni基催化剂反应性能的影响, 得到了反应性能和稳定性较好的Mg改性Ni基催化剂。
贵金属催化剂和镍催化剂具有较高活性, 碱土和稀土金属氧化物助剂可抑制镍催化剂上的积炭反应。由于甲烷部分氧化过程反应速率极快, 空速很高, 单位催化剂表面的放热量很大, 控制反应温度、避免催化剂床层飞温已成为反应器放大的又一关键问题; 此外, 原料气CH4/O2=2在高温下反应存在爆炸危险。对此, 研究人员从反应器、新工艺开发等方面进行了研究。中国科学院大连化学物理研究所采用含有氧分布器的固定床反应器, 以改性Ni-Al2O3为催化剂, 取得了较好效果[54, 55]; 中国石油大学则采用固定床两段法工艺将天然气低温催化燃烧与部分氧化联合, 避免了氧气浓度过高产生的安全隐患[56]; 重庆化工研究院开发了适用于薄层固定床反应器的催化剂体系[57]; Exxon公司开发完成了甲烷部分氧化制合成气的流化床新工艺, 新工艺采用甲烷、水蒸汽的混合气与氧气分开进料方式, 用水蒸汽消除了积炭的生成, 产物气必须快速降温冷却, 否则合成气在降温过程中将发生CO的歧化和甲烷化反应[58]。
影响甲烷催化部分氧化法生产成本的最大因素是从空气中分离制备纯O2, 所以开发与反应过程相结合的空气膜分离器, 形成反应与膜分离的耦合是该领域的一个重要研究方向。有研究者采用Ba0.5Sr0.5Co0.8Fe0.2O3复合物制成膜反应器, 实现了制氧和反应的耦合[59]。采用催化剂的晶格氧代替分子氧, 通过变价金属氧化物循环性还原-氧化, 实现甲烷晶格氧氧化制合成气反应是又一重要方向。由于反应中没有气相氧分子, 因而不受爆炸极限的限制, 可提高原料浓度而使产物易于分离, 降低了设备和分离费用; 同时以晶格氧作氧源还减少了催化剂表面的深度氧化, 提高了反应的选择性和经济性[60]。
6.3.3 生物质气化制合成气
生物质气化制合成气, 进而合成化工产品和液体燃料是一种效率高、成本低、无污染的可再生能源生产技术, 已成为研究热点, 是生物质转化利用技术中极具潜力的发展方向, 具有十分广阔的应用前景。
6.3.3.1 生物质气化基本原理和气化过程
生物质气化原理是指在一定热力学条件下, 借助空气、水蒸汽的作用, 使生物质高聚物发生热解、氧化还原、重整反应, 热解伴生的焦油进一步热裂化或催化裂化为小分子碳氢化合物, 获得含CO、H2和CH4气体[61]。
生物质气化过程根据反应温度和产物不同, 可分为干燥阶段、热解阶段、氧化还原阶段和净化阶段。
干燥阶段:脱除水分, 物料化学组成几乎不变。
热解阶段:生物质初步裂解炭化, 脱除挥发组分(主要是碳氢气体、H2、焦油、水蒸汽、CO、CO2), 得到固定碳和灰分。
氧化还原阶段:固定碳与O2反应生成CO、CO2和H2O, 放出大量热为碳深层气化提供能量。
净化阶段:产出气夹带少量水蒸汽、焦油、碱金属及氨, 须经冷却器、除尘器、水喷淋塔将其除去后, 方可输送至燃气设备。
生物质气化过程的总反应如式(6-4)所示, 同时包括式(6-5)~(6-11)所示反应:
CHxOy+O2+H2O→ CH4+CO+CO2+H2+H2O+C+tar (6-4)
2C+O2→ 2CO (6-5)
C+O2→ CO2 (6-6)
C+2H2→ CH4 (6-7)
CO+H2O→ CO2+H2 (6-8)
CH4+H2O→ CO+3H2 (6-9)
C+H2O→ CO+H2 (6-10)
C+CO2→ 2CO (6-11)
6.3.3.2 生物质气化过程研究现状
生物质气化技术早在18世纪就已出现, 但直到20世纪70年代, 受石油危机影响, 世界各国开始重新重视生物质能源的开发和研究。目前, 生物质气化技术的研究关键是增加可燃气中H2和CO浓度、燃气净化、减少二次污染以及提高转化率[62]。影响气化过程的关键因素包括气化反应器类型、气化介质、气化温度、气化压力、原料类型和性质以及催化剂的类型和用量等。
生物质气化反应器类型可分为固定床和流化床反应器, 固定床反应器又可分为上吸式、下吸式、横吸式以及开心式等, 流化床反应器又可分为鼓泡流化床和循环流化床。
固定床反应器气化的优点是设备简单, 成本低, 操作简便, 容易控制反应条件而且原料停留时间长, 碳转化率较高。上吸式固定床气化反应器出口气体温度较低(约250 ℃), 热效率高, 但气体中含有较多焦油, 后续处理困难。与上吸式固定床反应器相比, 下吸式固定床气化反应器得到的气体焦油含量非常低, 但出口气体温度较高(约800 ℃), 需回收气体带出的热量, 导致热效率较低。下吸式固定床气化反应器生产的气体约含有20%~28%的CO和约12%的H2, 含有少量焦油。固定床气化反应器投资少, 操作简单, 但处理速率慢, 生产能力低。
流化床气化反应器加强了物料之间的热传递, 从而提高了反应速率和转化效率, 具有物料处理量大、传热传质性能好等优点。此外, 流化床炉内温度较高且恒定, 加强了焦油裂解, 可燃气中焦油含量较少。但燃气中含有较多灰分, 需进一步处理。鼓泡流化床流化速率慢, 适合颗粒较大生物质原料, 须增加热载体, 固体颗粒带出较少; 循环流化床流化速率相对较高, 从流化床中携带出的颗粒在通过旋风分离器收集后重新送入炉内进行气化反应, 气化速率快, 适用于较小生物质颗粒。
生物质气化介质主要包括空气、氧气、水蒸汽、空气-水蒸汽以及氧气-水蒸汽。用空气作为气化介质具有成本低和操作可行性强优点, 但得到的气体热值较低[(4~7) MJ· m-3], H2体积分数只有8%~14%, 而且气化的气体含有大量N2, 需净化处理, 不适合后续合成; 用纯氧气化能避免N2带入, 可制取中热值[(10~12) MJ· m-3]气体, 具有工艺简单, 技术成熟, 运行稳定等优点, 得到的燃气适合用于合成, 但需专门制氧装置, 投资成本较高, 适用于大规模液体燃料合成工厂。采用水蒸汽作气化介质能够大量增加合成气中的H2含量, 反应(6-8)~(6-10)均需水蒸汽参加, 从反应平衡角度看, 水蒸汽越多越有利于H2的产生, 但水蒸汽过多会导致炉内温度下降, 从而需要外部供热。近年来, 用纯水蒸汽作为气化介质的气化技术得到广泛关注, 水蒸汽气化得到的气体热值较高[(12~20) MJ· m-3], 而且富含H2和CO, 适合作为合成气合成液体燃料[63, 64]。
气化温度是生物质气化过程中重要的工艺参数, 对产出气的种类、组成分布、热解速率和反应的热量变化有很大影响。随温度升高, 气体产率增加, 反应速率增大, 对产品气组成的影响则随实验条件变化而有所不同。乌晓江等[65]利用沉降式加压气化炉考察气化的影响因素时发现, 高温有利于气化反应向吸热方向进行, 在(1 327~1 427) ℃碳转化率高达90%, 合成气产量随氧碳比增加, 呈现先增后减的变化趋势。Song T等[66]研究串行流化床制氢实验中得出高温利于合成气生产和焦油分解, 在(720~920) ℃, 随温度升高, CO含量显著增加, 820 ℃时, H2产率和碳转化率达到最大值。Kumar A等[67]发现, 当气化温度由750 ℃增加到850 ℃时, 碳转化率、气体含热量以及H2所占比例有所增加。Gupta A K等[68]观察到在超过800 ℃的条件下, 当水蒸汽与生物质的质量比从0.5增加到1.0时, H2含量明显增加。Gonzá lez J F等[69]在研究空气气化过程中发现, 当温度由700 ℃增加到900 ℃时, H2和CO含量明显增加, 而甲烷和CO2含量有所下降, 提高反应温度有利于制取富含CO和H2的合成气。
气化压力对生物质气化有重要影响, 提高压力可在保持生产能力的条件下, 减小气化炉体积和后续处理设备尺寸; 此外, 加压气化生产压缩燃气可直接带压参与后续重整变换过程。从合成气角度看, 加压气化缺点在于提高压力使反应(6-7)和(6-9)向生成甲烷方向移动, 导致甲烷以及其他碳氢化合物含量有所上升, 为后续重整增加难度。黄海峰等[70]在生物质加压气化试验中观察到, 随压力升高, 甲烷等烃类气体含量呈上升趋势。
生物质气化过程会产生焦油等难以直接利用的物质, 不仅造成能量浪费, 还会影响系统正常运行。因此, 研究开发能够降低焦油产量的催化剂, 是生物质气化制合成气技术的一个关键问题, 也是研究热点。目前, 生物质气化除焦油最常用催化剂是Ni基催化剂。Magrini-Bair K A等[71]以90%α -Al2O3为载体, 负载质量分数分别为5.0%MgO、8.0%NiO和3.5%K2O得到的催化剂具有较好的焦油裂解效果, 在800 ℃下, 焦油裂解率可达90%以上, 其中载体α -Al2O3粒径在(100~400) μ m, 其抗磨损能力强, 经过48 h连续实验, 粒径分布无明显变化。Li C等[72]以铝酸钙为载体, 通过浸渍法负载硝酸镍制成的Ni基催化剂也可用于生物质气化制备富氢合成气, 在温度为650 ℃、气固比为2.1和时空速率为8.9 (kg· h)· m-3的条件下进行焦油裂解, 焦油转化率可达99%以上, H2产率可达80%, CO选择性可达63%。此外, 采用浸渍法经(400~500) ℃处理得到的纳米Ni基催化剂, 对于提高H2产率和焦油转化率效果明显[73]。Ni基催化剂的主要问题是失活比较严重, 其中由于H2S中毒而使Ni活性位点减少是导致催化剂失活的主要原因, 同时烧结导致Ni晶体变大以及炭化现象也可能造成催化剂失活[74]。Rh基催化剂也是一种有效焦油裂解催化剂, Colby J L等[75]在气化炉温度850 ℃、压力0.1 MPa条件下, 采用以α -Al2O3为载体、负载Rh制备的催化剂, 可使焦油转化率达到50%。Tomishige K等[76]以SiO2为载体, 负载Rh和CeO2制备的催化剂, 用于催化焦油裂解和生物质气化, 在温度650 ℃、压力0.1 MPa、生物质进料量85 mg· min-1和空气流量50 m3· min-1条件下, 碳转化率达99%以上, 可得到CO产量为2 254 μ mol· min-1, H2产量为2 061 μ mol· min-1的合成气。Rh基催化剂在使用中的最大问题是催化剂磨损和失活。除Ni基和Rh基催化剂外, 生物质气化制合成气中, Ru、Zr、Pt等重金属对焦油去除也有一定效果, 但目前研究较少[77]。不管采用哪种催化剂, 在合成气制备过程中普遍存在焦油转化率较低问题, 虽然有些催化剂可实现理想焦油转化率, 但成本较高, 因此研究开发催化效率高且价格低廉的新型焦油裂解催化剂是生物质气化制合成气技术一个亟待解决的关键问题。
6.3.3.3 未来生物质气化技术的研究方向
近30多年来, 人们对生物质气化技术进行了大量实验研究, 但生物质气化制合成气的研究大多为实验室研究和小规模中试研究, 大型生产工艺和配套设备还有待进一步开发。目前多数生物质气化制合成气技术与传统技术相比仅有社会环境效益, 无经济竞争优势, 这使得该技术的工业化生产受到限制。目前生物质气化制合成气技术研究仍有很多问题急需解决, 主要体现在:(1) 生物质气化反应器对各类生物质或混合生物质原料气化试验的通用性不强; (2) 现有生物质气化技术所得到的产气成分不符合化学品合成技术的要求。产气中氢碳比一般较低, 达不到甲醇、乙醇等化学品合成的理论比例, 而且产气中的CO2、CH4含量较高, 影响后期液体燃料合成; (3) 生物质气化制合成气过程中会产生大量难以利用的焦油, 影响产气效果和系统运行。
针对上述亟待解决的问题, 生物质气化技术未来的研究将主要集中在如下几个方面:(1) 以增强反应器适应生物质原料通用性为目标的生物质气化反应器的改进和研发; (2) 以提高有效产气成分(H2和CO)产率为目标的生物质气化技术的研究; (3) 以尽量减少气化过程中的焦油产量为目标的改善气化条件研究和新型廉价高效焦油裂解催化剂的设计和研发。
6.4 合成气转化利用
6.4.1 合成气转化利用概述
合成气转化利用是指合成气(或合成气与化学品)反应制备(或生产)液体燃料和化工产品的过程。作为合成气中枢的两大核心任务之一, 合成气转化利用在合成气中枢中起着非常重要的作用:可为合成氨厂提供H2/N2原料气, 为炼油厂提供高品质柴油、润滑油调和组分以及宝贵的H2, 为石油化工厂提供石脑油、甲醇/二甲醚, 进一步转化为低碳烯烃, 同时还可为发电厂提供合成气(H2/CO)或H2实行涡轮机发电或燃料电池发电, 同时通过“ 多联产” 提供化学品; 可提供合成天然气(SNG), 补充天然气资源不足, 可大大减轻大量烃类、长距离输送的沉重负担。
6.4.2 合成气转化制含氧化合物
含氧化合物是指由合成气生成的含有C、H、O的有机化合物, 包括醇、醚、酯、醛和酸等。这些含氧化合物既可作为燃料, 如甲醇、乙醇和二甲醚等, 又可作为重要的化工品原料, 如低碳醇、乙二醇和丙三醇等。
6.4.2.1 甲 醇
甲醇是极为重要的有机化工原料和清洁液体燃料, 是合成气转化的基础化学品。合成气合成甲醇的主要反应如式(6-12)所示, 同时合成气中的少量CO2也发生加氢反应生成甲醇[如式(6-13)所示], 反应的同时还发生水气变换反应[如式(6-14)所示]:
CO+H2=CH3OH
△ Gθ =-113.791+0.243 4T, kJ· mol-1 (6-12)
CO2+H2=CH3OH+H2O
△ Gθ =-78.603+0.212 1T, kJ· mol-1 (6-13)
CO+H2O=CO2+H2
△ Gθ =-35.503+0.032 1T, kJ· mol-1 (6-14)
合成甲醇涉及的主要反应[式(6-12)~(6-14)]均为放热反应, 低温有利于反应进行; 同时甲醇合成又是分子数减少的反应, 增加压力有利于甲醇合成。从式(6-12)~(6-14)可知, 在热力学上CO+H2合成甲醇比CO2+H2合成甲醇更有利。
工业上合成甲醇的发展, 很大程度上取决于催化剂的研发及其相应操作条件的保证, 不同的生产工艺就有不同的催化剂及其相应的反应器来实现。目前, 虽然有多种催化剂可以催化合成气合成甲醇, 但工业上大规模应用的只有锌-铬基催化剂和铜基催化剂。锌-铬基催化剂(ZnO/Cr2O3)是德国BASF公司于1923年首先开发研制成功, 其活性温度较高, 适宜的温度为(320~400) ℃, 为获取较高的合成气转化率, 其操作压力也较高, 通常为(25~35) MPa。锌-铬基催化剂具有较好的耐热性、抗毒性、机械强度、使用寿命, 但活性温度与操作压力高, 致使动力消耗大, 设备复杂, 产品质量差。随着英国ICI公司在1966年将铜基催化剂成功应用于工业低压合成甲醇过程, 锌-铬基催化剂逐渐淡出合成甲醇市场。目前, 世界上的甲醇厂全部采用铜基催化剂。铜基催化剂低温活性高, 适宜操作温度(230~310) ℃, 压力(5~15) MPa[78]。
铜基催化剂的主要组分为CuO/ZnO/Al2O3或CuO/ZnO/Cr2O3, 其中CuO/ZnO是催化剂的活性组分, Al2O3或Cr2O3组分则明显提高了催化剂的抗老化能力。由于Cr对人体有害, 工业上普遍采的甲醇合成催化剂为CuO/ZnO/Al2O3。该催化剂研究开发机构主要有英国ICI、美国UCI、德国Lurgi和BASF、日本MGC、丹麦Topsϕ e公司和国内的西南化工研究院、西北化工研究院、南京化学工业集团研究院等[79]。
催化剂上合成甲醇的反应机理已被人们进行了大量研究, 但由于反应体系的复杂性和各反应之间的强烈耦合作用, 目前在许多问题上仍未达成共识, 存在诸多争论, 这些争论主要集中在甲醇产物中碳的来源问题(即甲醇中的碳是来源于CO还是CO2)、CO2作用以及Cu-ZnO催化剂的活性中心。而基于上述问题的不同认识, 产生了不同反应机理, 主要有以下三种观点:(1) CO为碳源机理。认为CO是甲醇产物中碳的唯一来源, CO2的存在仅仅起到稳定晶格(Cu+)作用, 即利用其弱氧化性保证低价铜不被过渡还原, 或通过逆变换反应转化为CO, 再经过CO在催化剂表面吸附生成中间物而合成甲醇, Cu+是反应的主要活性中心[80, 81, 82]; (2) CO2为碳源机理。认为CO2是甲醇产物中碳的来源, CO的作用则是作为一种还原剂使活化的Cu表面得以再生或用于清除CO2加氢中所产生的吸附氧, Cu0是反应的活性中心[83, 84, 85]; (3) CO和CO2双碳源机理。认为CO和CO2都可以加氢生成甲醇, 它们在不同能量状态的铜活性中心上, 以不同形式被活化[86, 87, 88]。这种更为折中的双碳源机理自20世纪80年代以来似乎越来越多地为人们所接受。
6.4.2.2 二甲醚
二甲醚不仅是合成气经甲醇制汽油和低碳烯烃的中间体, 而且是多种化工产品的重要原料。二甲醚在应用于气雾剂和制冷剂方面是氟氯烃的理想替代品, 而作为洁净液体燃料, 二甲醚的应用前景更为广阔。目前, 合成气合成二甲醚主要包括两种方法, 即合成气经甲醇合成二甲醚的两步法和合成气直接合成二甲醚的一步法。
两步法指的是合成气首先合成甲醇, 然后甲醇脱水生成二甲醚, 该法相对来说较为成熟。其中, 甲醇脱水生成二甲醚反应如式(6-15)所示:
2CH3OH=CH3OCH3+H2O
△ Hθ =-23.4 kJ· mol-1 (6-15)
该反应是放热反应, 低温有利于二甲醚合成。目前, 最常用工艺为甲醇气相脱水制二甲醚, 该工艺可连续生产二甲醚, 具有规模大、操作容易控制、无腐蚀、无污染物和废弃物排放等特点。该过程甲醇转化率高, 二甲醚选择性好。其关键是催化剂的研制, 常用的催化剂是酸性脱水组分如HZSM-5、γ -Al2O3等。
甲醇脱水合成二甲醚的反应机理主要在Al2O3和HZSM-5催化剂上进行研究, 得出的结论也不一致, 其主要争论之处在于甲醇是以什么状态吸附在催化剂表面; 催化剂吸附甲醇活性位的性质以及二甲醚形成的主要途径。如Padmanabhan V R等[89]描述了Al2O3上甲醇脱水制二甲醚历程, 认为甲醇在Al2O3上吸附有两种形式:以分子状态吸附在酸性位上与以解离态吸附在碱性位上, 吸附态的甲醇分子与解离后形成的甲氧基反应形成二甲醚。Rinaldo S S等[90]认为二甲醚形成有两种途径:一种是吸附的甲醇分子与甲氧基之间发生反应, 另一种则是两个吸附的甲氧基之间反应。低于280 ℃, 前者占优势; 高于280 ℃, 后者占优势。解峰等[91]则认为甲醇在氧化铝表面的不同L酸性位以分子态和解离态吸附, 分子态甲醇与邻近的解离吸附态甲醇(甲氧基)相互作用生成二甲醚。
一步法指的是合成气在具有甲醇合成和甲醇脱水的双功能催化剂上直接合成二甲醚。该反应过程实质上是甲醇合成、甲醇脱水和水气变换反应的耦合, 主要反应式如(6-16)~(6-18)所示:
CO+2H2=CH3OH
△ Hθ =-90.4kJ· mol-1 (6-16)
2CH3OH=CH3OCH3+H2O
△ Hθ =-23.4kJ· mol-1 (6-17)
CO+H2O=CO2+H2
△ Hθ =-40.9 kJ· mol-1 (6-18)
总反应如反应(6-19)~(6-20)所示:
2CO+4H2=CH3OCH3+H2O
△ Hθ =-205.2 kJ· mol-1 (6-19)
3CO+3H2=CH3OCH3+CO2
△ Hθ =-246.3 kJ· mol-1 (6-20)
反应(6-19)是不发生水气变换反应时的二甲醚合成, 反应(6-20)是发生水气变换反应时二甲醚的合成。一般来说, 甲醇合成催化剂同时也催化水气变换反应, 所以总的二甲醚反应介于反应(6-19)和(6-20)之间。
反应(6-19)和(6-20)均为放热反应和分子数减少反应, 降温和增压有利于原料转化和二甲醚生成。
同两步法相比, 该过程具有流程短、投资省、能耗低等优点, 同时可获得较高单程转化率, 因此, 合成气直接制二甲醚反应自20世纪80年代起一直受到人们关注, 对其进行了大量研究, 该过程研究的关键是多功能催化剂的研制, 目前研究较多的是由Cu/ZnO/Al2O3甲醇合成活性组分和γ -Al2O3、HZSM-5等酸性脱水组分组成的双功能催化剂, 如何实现两种活性组分操作条件的匹配, 充分发挥它们的协同作用, 保持高催化性能和催化稳定性, 是合成气一步法制二甲醚双功能催化剂的研究热点。
关于合成气一步法合成二甲醚的机理, 普遍认为是合成气在Cu基催化剂表面生成甲醇, 然后甲醇转移到固体酸催化剂表面发生脱水反应生成二甲醚。在双功能催化剂上, 甲醇合成与甲醇脱水均在催化剂表面形成甲氧基自由基, 当两种活性位接触足够紧密时, 甲基自由基可从甲醇合成活性位直接跃迁到甲醇脱水酸性位, 这样可使部分反应不经过甲醇中间体而直接转化为二甲醚[92]。
6.4.2.3 乙 醇
乙醇是重要的基本化工原料, 在工业上广泛用作溶剂、消毒剂和有机合成原料, 同时乙醇在作为燃料和燃料添加剂方面具有较为广泛的用途。乙醇的工业生产有天然原料发酵法和化学合成法两大类。发酵法是生产乙醇的经典方法, 通常以糖类、淀粉和纤维素等碳水化合物为原料, 经发酵使双糖、多糖转化为单糖, 并进一步转化为乙醇。化学合成法指的是以乙烯为原料的水合法。目前正研究开发碳一化工路线合成乙醇的方法, 其中包括合成气直接合成法。
合成气直接合成乙醇过程的主要反应如式(6-21)所示:
2CO+4H2=C2H5OH+H2O
△ Hθ =-253.6 kJ· mol-1 (6-21)
由上式可见, 合成气直接转化制乙醇是一个强放热且容易进行的反应, 低温有利于反应的进行; 同时合成气直接合成乙醇反应是分子数减少的反应, 增加压力有利于乙醇合成, 合成乙醇需要H2/CO物质的量比为2:1。
由于受多种因素影响, 合成气直接合成乙醇的反应总伴随有副反应发生, 导致产生甲烷、C2~C5的烷烃和烯烃等, 同时伴随着水气变换反应, 产生大量CO2。因此, 通过研发合适的催化剂来抑制副反应发生是提高产物乙醇选择性和产率的重要途径。
目前, 关于合成气合成乙醇催化剂的研究, 可分为贵金属催化剂和非贵金属催化剂两类。美国联碳公司最先进行该过程研究, 采用Rh/SiO2催化剂研究合成气选择合成乙醇、乙醛和醋酸等C2含氧化合物, 但催化剂活性低并副产大量甲烷。为改善催化剂性能提高目的产物的选择性, 研究了各种助剂对催化性能的影响, 发现加入Fe、Ir、Ti 等金属增强加氢能力, 加入Mn、Sc、V、Zr 等金属增强CO 离解能力, 加入K、Li 等金属抑制加氢能力[93]。Burch R等[94]采用2%Rh-10%Fe/Al2O3催化剂, 在压力1 MPa下, 由合成气选择性合成乙醇, 选择性最高达50%。中国科学院大连化学物理研究所已在重庆垫江建成30 t· a-1 乙醇中试装置, 该装置以天然气为原料, 经由合成气生产乙醇, 经1 030 h连续运转后催化剂时空产率达315 g· (kg-cat· h)-1 , 产物中C2+含氧化合物的选择性达73%[95]。鉴于合成气直接合成乙醇反应中产品选择性较低的问题, 研究者提出了两步转化即复合体系催化剂方案, 首先用Rh-Mn-Li/ SiO2催化剂合成C2含氧化合物, 不分离产物, 然后再用Cu-Zn/SiO2 催化剂使醋酸和乙醛转化为乙醇, 乙醇选择性达82%, 时空产率为230 g· (L· h
合成气直接制乙醇的机理研究认为, 在铑基催化剂上合成气直接制乙醇反应大体分为以下步骤进行:首先, H2、CO被催化剂吸附, 接着吸附在催化剂上的CO自身分解, 然后被氢化, 并在催化剂表面形成一种碳氢化合物(CHx)ad(x=2或3); 这时未分解CO插入到Rh-C键(CHx物种中的C)中, 同时被氢纯化后得到烯醇中间体; 烯醇中间体会与Had原子反应生成乙醇[98]。大多数研究者认为CO在Rh基催化剂上的吸附是合成气制乙醇反应的速控步骤。在合成乙醇过程中, 常常伴随有多种副反应发生, 如CO分解形成的Oad原子可能与CO反应生成CO2; (CHx)ad物种可能会被氢化形成甲烷; 烯醇中间体会与吸附的H原子和CO反应生成C2+含氧化合物[99]。
6.4.2.4 低碳醇
低碳醇是以合成气为原料, 在催化剂作用下合成的C1~C5 醇类混合物总称。低碳醇可以混合醇的形式直接作为动力燃料, 还可作为燃料添加剂; 也可分离得到单独的醇类作为化工原料。
合成气合成低碳醇反应的化学式及其反应的吉布斯自由能变△ Gθ 与温度的关系如式(6-22)和(6-23)所示:
nCO+2nH2=CnH2n+1OH+(n-1)H2O (6-22)
△ Gθ =-38.385n+11.100+(6.002n-0.150)T/100, kJ· mol-1 (6-23)
由上式可知, 合成低碳醇的过程属于分子数减少反应, 高压有利于反应的进行; 同时低温有利于低碳醇的合成, 但温度太低会使反应速率降低, 因此在低温下采用合适的催化剂体系, 是该过程达到高转化效率的关键。
合成气合成低碳醇的催化剂体系根据催化剂活性组分的性质和反应类型主要分为以下几类催化剂[100]:(1) 改性高温甲醇合成催化剂体系。该类催化剂由高温甲醇合成催化剂(Zn-Cr氧化物)加入碱性助剂改性制得, 反应条件为温度约400 ℃, 压力(15~32) MPa。一般认为具有非化学计量性质的Zn-Cr类尖晶石结构起活性组分作用, 产物主要是甲醇、异丁醇、乙醇和正丙醇, 产物中异丁醇占相当大比例; (2) 改性低温甲醇合成催化剂体系。该类催化剂是通过向低温铜基甲醇合成催化剂中加入碱性助剂提高低碳醇的选择性, 产物中除甲醇外, 以乙醇、正丙醇、正丁醇和更高碳数醇为主, 反应条件为温度(300~350) ℃, 压力约10 MPa; Cu/ZnO是反应的主要活性组分; (3) 改性F-T合成催化剂体系。将甲醇合成与F-T合成催化剂结合, 加入碱金属助剂, 可缓和F-T合成催化剂的还原作用, 提高产物中醇类的选择性, 该类催化剂上反应产物主要为遵循Andersen-Schulz-Flory(ASF)分布的C1~C5正构醇; (4) MoS2催化剂体系。该类型催化剂反应条件温和(约300 ℃和10 MPa), 具有较强的抗硫中毒作用, 但产物中含有大量烃和CO2使得低碳醇选择性不高。目前合成气合成低碳醇催化剂的研究重点在于探索活性中心的最佳匹配、构效关系及合成低碳醇的选择性规律等, 旨在提高低碳醇合成过程的单程转化率、CnH2n+1OH (n=2~5)选择性和醇产率[101]。
关于合成气合成低碳醇的机理在Cu基催化剂上研究较多, 多数认为合成低碳醇的机理涉及碳链增长, 碳链增长的关键是CO的解离吸附, 作为活性中心的Cu在(200~300) ℃对CO的吸附为非解离吸附。有两种方法可使CO解离, (1) 提高反应温度到300 ℃以上, 使其易于解离, 但温度不易过高, 以免生成大量的烷烃副产物; (2) 设法削弱活性中心铜上吸附CO的碳氢键。加入碱金属, 可降低金属铜的电子逸出功, 即增强了铜的电子给予能力, 使CO得到更多的反馈电子, 因而减弱了碳氢键, 导致CO解离, 为碳链增长创造条件[102, 103]。
6.4.2.5 乙二醇
乙二醇是最简单也是最重要的脂肪族二醇, 它是一种重要的大宗化工原料, 可用于生产聚酯纤维、防冻剂、不饱和聚酯树脂、乙醇胺等产品。目前工业上合成乙二醇的主要方法是采取乙烯出发的环氧乙烷水合法, 该法的经济效益受石油价格影响较大, 开发和改良乙二醇合成路线已成为研究热点。以合成气为主要原料的乙二醇合成工艺, 因其符合我国能源资源分布特点, 原料来源广泛和低廉, 技术经济性高而引起愈来愈多的重视。
乙二醇的合成气合成法分为直接合成法和间接合成法。直接合成法采用合成气直接合成乙二醇, 间接合成法则是合成气经乙烯、草酸酯、甲醇、甲醛等中间产物合成乙二醇。
合成气直接合成乙二醇的反应为:
2CO+3H2=HOCH2CH2OH (6-24)
从形式上看, 由合成气直接合成乙二醇符合原子反应经济性的要求, 是一种最为简单和有效的乙二醇合成方法, 即使反应选择性和转化率较低, 也具有很大的实际应用价值。目前合成气直接制乙二醇研究的关键是如何改进和提高催化剂的活性和选择性。在研究的合成气直接制乙二醇的催化剂体系中, 两种催化剂的性能相对较好, 即四烷基膦和胺改性的铑催化剂和咪唑改性的钌催化剂[104]。虽然迄今直接法还未实现工业化, 但只要有所突破, 使反应在较温和条件下进行, 该过程将非常有竞争力。
相对于直接法, 合成气间接法合成乙二醇, 如甲醇/甲醛法、草酸酯法取得的进展较多, 特别是草酸酯法合成乙二醇, 已发展成为离工业化合成乙二醇最近的一条工艺路线。
甲醇/甲醛法第一步甲醇、甲醛的合成工艺技术成熟, 建有百万吨级的生产装置。虽然从甲醇、甲醛出发, 合成乙二醇的工艺路线有很多, 但目前离工业化实施还有一段距离。该法制乙二醇目前的几个主要研究方向包括甲醇二聚法、二甲醚氧化法、甲醛二聚法、甲醛羰基化法和甲醛氢甲酰化法[105]。
草酸酯法(也称为氧化偶联法)合成乙二醇主要包括两个步骤, 即CO通过氧化偶联制草酸酯, 然后再加氢合成乙二醇。液相合成草酸酯早在1968年就由美国Union Oil公司申请专利。1978年, 宇部兴产公司和美国UCC公司联合开发了合成草酸二酯的新工艺路线。该工艺是在亚硝酸酯的存在下, 采用以活性炭为载体的钯催化剂, 用CO、O2和正丁醇反应, 反应温度90 ℃, 压力9.8 MPa, 生成草酸二丁酯[106]。1977年日本宇部兴产公司提出常压气相合成草酸酯技术, 以Pd/Al2O3为催化剂, 在温度(80~150) ℃和压力0.5 MPa条件下, 草酸二甲酯收率98%[107]。同时, 在气相法制草酸二甲酯基础上, 宇部兴产公司采用Cu-Cr催化剂, 开发出气相法草酸二甲酯加氢还原制乙二醇技术, 乙二醇选择性为95%[108]。1986年美国ARCO公司首先申请了草酸酯加氢制乙二醇专利, 开发了Cu-Cr催化剂, 采用Cu-Cr催化剂, 在3.0 MPa压力下, 催化剂最长运转466 h, 乙二醇收率为95%[109]。同年, 宇部兴产公司与UCC公司联合开发出Cu/SiO2催化剂, 乙二醇收率97.2%[110]。国内对气相法合成草酸酯进行深入研究的有多家单位, 包括中国科学院福建物质结构研究所、天津大学、华东理工大学、上海焦化有限公司和浙江大学等。国内对催化剂的研究多以α -Al2O3为载体, 以Pd为活性组分, 仅催化剂助剂有差别, 开发的催化剂均取得了较好效果。国内的多家单位同时开发了草酸酯加氢的Cu基催化剂, 也取得较高的催化性能。Cu基催化剂反应条件温和、活性高、乙二醇选择性好, 但是抗烧结能力差、机械强度低, 并且容易在催化剂表面形成草酸铜和聚酯, 使得催化剂的稳定性下降、寿命缩短[111]。目前, 关于合成气经草酸酯合成乙二醇, 草酸酯的生产技术已工业化, 研究重点集中在草酸酯催化加氢的催化剂和工艺技术改进, 一旦取得突破, 就可实现合成气工业化生产乙二醇。
6.4.3 合成气转化制烃燃料
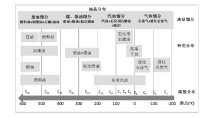
作为燃料的烃类, 烃中所含碳原子数不同, 燃料用途也有所不同。如CH4是合成天然气的主要组分、C2~C4烃可用作民用燃气、C5~C12烃是汽油的主要组分、C10~C16烃是航空燃油的主要馏分、C15~C20烃是柴油的主要组分、C18~C22是润滑油的主要组分等(如图6-3所示)[112]。这些作为燃料的烃类, 既可来源于石油加工, 也可从煤、天然气、生物质等资源经合成气制备。随着石油资源的逐渐减少, 替代石油资源制备烃类燃料就显得尤为重要, 而合成气转化制备烃燃料作为替代油资源制备烃燃料的重要步骤, 在能源领域一直扮演重要角色。合成气转化制备烃燃料过程可分为合成气直接合成烃和合成气经醇醚间接合成烃燃料。
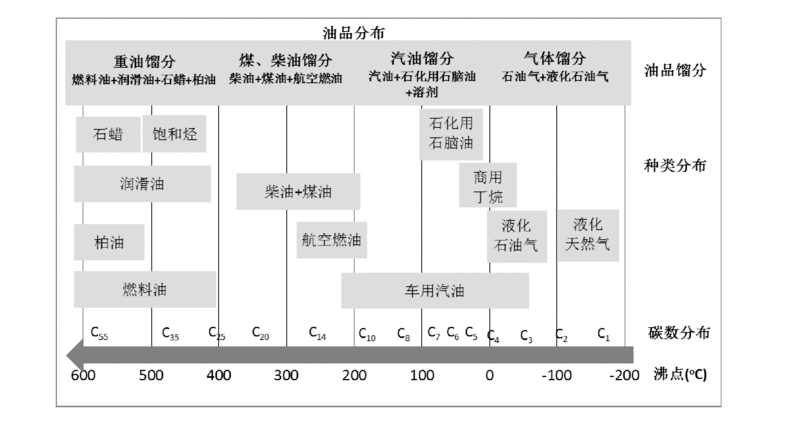 | 图6-3 作为燃料烃产品的碳数、沸点分布及应用 |
6.4.3.1 合成气直接转化制烃燃料
合成气直接转化制烃燃料最著名的过程是F-T合成, 是以合成气为原料在催化剂和适当条件下合成液态烃或碳氢化合物的工艺过程。该过程由德国化学家Fischer和Tropsch共同开发, 是气体液化技术的一个关键组成部分, 通常是从煤、天然气或生物质生产合成润滑油与合成燃料。F-T合成作为低硫柴油燃料的来源而得到间歇性关注, 用以解决基于石油烃类的供应或成本问题。F-T合成烃燃料的主要反应式为:
nCO+(2n+1) H2→ CnH2n+2+nH2O
△ rHm=-139.958n-35.337+(-28.44-51.44n)T/1000+(1.403+7.771n)/2× 10-5T2+(1.226-2.12n)/3× 10-8T3-(0.7645+0.617n)/4× 10-11T4 , kJ· mol-1 (6-25)
nCO+2nH2→ CnH2n+nH2O
△ rHm=-139.958n+82.240+(2.51-51.44n) T/1000+(-2.765+7.771n)/2× 10-5T2+(1.478-2.12n)/3× 10-8T3-0.617n× 10-11T4, kJ· mol-1 (6-26)
CO+H2O→ CO2+H2
△ rHm=-39.658-6.17T/1000+9.364/2× 10-5T2-10.827/3× 10-8T3+4.1115/4× 10-11T4, kJ· mol-1 (6-27)
上述反应式中, 式(6-25)和式(6-26)分别为生成直链烷烃和α -烯烃的主反应, 式(6-27)为F-T合成过程中伴随的水气变换反应。从反应焓变△ rHm与温度T(K)的函数关系可推断在温度(373~773) K条件下, F-T合成反应的反应焓皆小于0, 是放热反应, 低温有利于反应进行, 同时生成烃类燃料的反应为分子数减少反应, 高压有利于产物烃类燃料的生成。
实际上, F-T合成反应的产物分布和热力学平衡差异很大, 这是因为受控于动力学条件造成的, 也就是受催化剂和反应工艺参数的影响, 其中催化剂特别重要。当采用合适催化剂时, 可大大减少非目的产物(如甲烷)的生成[113, 114]。
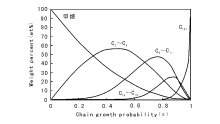
传统F-T合成所得产品烃的碳数分布通常遵循Anderson-Schulz-Flory(ASF)规律, 即有一定的链增长几率, α (α =Cn+1/Cn), 随α 值升高, 产物分布趋重, 即选择性趋向于重烃, 如图6-4所示[115]。
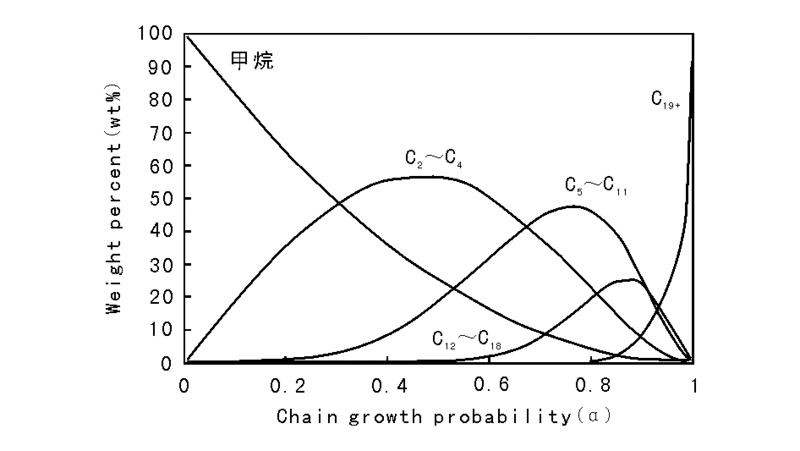 | 图6-4 F-T合成的烃产品分布 |
从图6-4可以看出, 只有甲烷和高分子蜡有较高的选择性, 其余馏分烃都有选择性极限:C2~C4为60%, C5~C11为48%、C12~C18为25%。因此F-T过程只能得到混合烃产物, 选择性差是F-T合成过程的一个显著特征。为做到选择性合成烃类产品, 许多研究者正致力于开发不服从ASF烃产品分布的催化剂和工艺。Shell、Exxon、Syntroleum、Sasol等公司都将催化剂的研究开发作为GTL技术中的一个重要环节。如为降低气体烃的生成率, SMDS(Shell Middle Distillate Synthesis)及SSPD(Sasol Slurry Phase Distillate)工艺均选择了可获得较高链增长几率的催化剂, 合成产品主要是高分子的蜡, 然后经缓和加氢裂化得到馏分油产品[116, 117]。应当强调指出, F-T合成所得产品不含硫、氮、芳烃, 主要是直链烷烃及α -烯烃, 汽油辛烷值低而柴油十六烷值高。
F-T合成烃催化剂的活性金属组分中以Fe、Co、Ni、Ru、Rh最为活泼, 这些元素的链增长概率为:Ru> Fe~Co> Rh> Ni。一般认为Fe和Co具有工业价值, Ni有利于生成甲烷、Rh易于生成含氧化合物、Fe则易于生成大量烯烃和含氧化合物, Ru、Co易于合成长链饱和烃[118]。这些元素在反应条件下以金属、氧化物或碳化物状态存在。
由于比较复杂的众多反应产物和研究者不同的侧重点, 人们对F-T合成机理的认识必然存在差异和争论。目前得到较大范围认可的经典F-T合成机理主要有碳化物机理、含氧中间体缩聚机理、CO插入机理和双中间体机理等[119]。普遍认同的观点是:CO在催化剂表面活性中心上的解离是F-T合成最基本的重要步骤, 弄清楚合成反应机理有助于解决反应的起始、链增长以及产物分布和动力学研究问题。研究表明, 在复杂的F-T合成反应体系中可能不存在单一反应机理, 或许F-T合成产物分布最终受几种反应机理共同作用(譬如CO在催化剂表面上同时进行离解与不离解吸附已成为事实), 只不过某种反应机理在反应中起着主要制约作用[120]。
合成气直接制烃类液体燃料的另一条技术路线是在由合成甲醇活性组分和金属改型分子筛组成的复合催化剂上实现合成气直接定向转化制燃料烃[121, 122, 123]。与F-T合成路线相比, 该路线的烃类产品分布不受F-T合成烃产品ASF分布规律的限制, 定向转化的目标产品烃可获得较高选择性, 同时产品烃中甲烷含量较低, 合成的产品烃以异构烷烃为主[3]。
该过程的主要反应式为:
2nCO+(n+1)H2=CnH2n+2+nCO2 (6-28)
该反应是放热反应, 低温有利于反应进行, 同时又是分子数减少反应, 增大压力有利于反应进行。目前该过程由于采用催化剂体系和反应条件的不同, 而得到不同的低碳烃馏分; 如Cu-ZnO/Pd-β 催化剂上可高选择性合成C3~C4烃, Cu-ZnO/Pd-SAPO-34催化剂可高选择性生成C2~C3烃, Cu-ZnO/Zn-ZSM-5催化剂可高选择性合成C5~C11烃等[124, 125, 126]。
该过程常用催化剂体系为甲醇合成催化剂如Cu-ZnO、ZnO-Cr2O3等, 金属改性分子筛如金属改性的HZSM-5、HY、Hβ 、HSAPO-5、HMCM-41等。目前关于催化剂上合成气转化制烃的机理研究不多, 得到大多数研究者认可的反应路径为:合成气首先合成甲醇(二甲醚), 然后发生甲醇(二甲醚)脱水反应生成烯烃, 烯烃加氢生成烷烃。日本北九州市立大学、中国科学院大连化学物理研究所等均在该方面进行了系统研究, 主要研究方向集中于复合催化剂稳定性的改进和相关反应机理研究[121, 122, 123, 124, 125, 126, 127, 128]。
6.4.3.2 合成气间接转化制烃燃料
合成气间接转化制烃燃料是指合成气经醇醚等中间化合物合成烃类燃料的过程, 代表过程是由来自合成气的甲醇制汽油的MTG过程(Methanol to Gasoline)。
MTG过程是Mobil公司开发的以甲醇为原料, 在一定温度、压力和空速下, 通过特定的催化剂进行脱水、低聚、异构等步骤转化为汽油烃的过程[129]。可用下式表示:
CH3OH→ CH3OCH3(+H2O) → C2 to C5 alkenes(+H2O) → 石蜡烃\芳烃\环烷烃 (6-29)
MTG过程是一放热过程, 反应的放热程度随产品烃分布情况而有所不同。因此, 控制和移除大量反应热是该类反应设计要考虑的一个重要因素。
MTG过程主要采用分子筛催化剂体系, 如HZSM-5催化剂用于MTG过程体现了以下特点: 选择性好, 活性高, 芳构化能力强; 产品分布优良, 以异构烷烃和芳烃为主, 同时含有少量的直链烷烃、烯烃和环烷烃; 在不含四乙基铅的情况下, 产物汽油的辛烷值可达90~95[130, 131]。
MTG过程在HZSM-5上的反应机理目前尚不十分清楚, 特别是关于第一个碳碳键的形成问题争议很大。有研究认为, 乙烯是最初产品, 形成的第一个碳碳键为乙烯键; 还有研究认为, 丙烯是最终产品, 形成的第一个碳碳键是丙烯键。不同硅铝比的HZSM-5分子筛催化甲醇制汽油的初始产物分析表明, 丙烯是主要初始产物, 支持了形成的第一个碳碳键是丙烯的假设[132]。
6.4.4 合成气转化制低碳烯烃
乙烯、丙烯等低碳烯烃是重要的基本有机化工原料, 是现代化学工业的基石, 其传统生产技术强烈依赖于石油资源, 随着石油资源的日趋减少, 发展以非石油资源为原料制取低碳烯烃等石化产品, 实施石油替代战略, 是关系到世界上许多国家经济长期稳定发展和能源安全的重大课题。非石油资源如煤、天然气、生物质等制取低碳烯烃的过程, 主要包括非石油资源的气化生产合成气, 再由合成气制取低碳烯烃, 而合成气制取低碳烯烃的过程又分为合成气直接合成低碳烯烃和合成气间接合成低碳烯烃。间接法工艺成熟, 已步入工业化阶段, 但从长远考虑, 合成气直接制取乙烯、丙烯的工艺比间接法更为经济[133]。
6.4.4.1 合成气直接转化制低碳烯烃
目前研究较多的合成气直接合成低碳烯烃过程为合成气经F-T过程合成低碳烯烃, 其工艺流程比间接法简单, 经济评价也较合算, 包含许多平行反应和顺序反应, 主要反应式为:
CO+2H2→ (-CH2-)+H2O
△ H(227 ℃)=-164.7 kJ (6-30)
CO+H2O→ H2+CO2
△ H(227 ℃)=-39.7 kJ (6-31)
2CO+H2→ (-CH2-)+CO2
△ H(227 ℃)=-204.4 kJ (6-32)
CO+3H2→ CH4+H2O
△ H(227 ℃)=-214.4 kJ (6-33)
2CO→ C+CO2
△ H(227 ℃)=-133.8 kJ (6-34)
这些反应既相互竞争又相互依存。比较(6-30)和(6-32)的反应焓变可知, CO2比H2O更容易生成。由上述反应的吉布斯自由能变与反应温度的关系可推知:在(200~400) ℃, 甲烷化和积炭的生成在热力学上是有利的, 低碳烯烃的生成不利。此外, 低碳烯烃生成的反应为分子数减少反应, 增加压力有利于反应进行。
合成气直接合成低碳烯烃的关键技术是催化剂, 要求催化剂能够限制碳链增长、抑制甲烷生成、阻止反应生成的低碳烯烃发生二次反应, 并具有较高的催化活性[134]。国内外相继在催化剂体系、制备方法及改性等方面做了很多研究, 催化剂种类也随之丰富起来。由于第Ⅷ 族元素对CO和H2良好的吸附作用, F-T合成催化剂一直将其作为催化剂的主要活性组分, 尤其以铁、钴、镍、钌研究最多, 分别呈现出铁系、钴系、镍系等系列催化剂体系。但从实际生产来看, 铁价廉易得且稳定, 相同转化率下较镍生成的甲烷少、烯烃多。因此, 铁系催化剂是合成气直接制取低碳烯烃较常用的催化体系, 关于催化剂上合成气直接制低碳烯烃的研究主要集中在改进催化体系的低碳烯烃选择性、反应稳定性和反应结果的可重复性、催化剂上反应热的及时移除等, 研究较为活跃的单位有中国科学院大连化学物理研究所、中国科学院山西煤炭化学研究所、天津大学、太原理工大学和华东理工大学等, 但目前关于该方面的研究仍处于实验室研发阶段[135]。
合成气经F-T路线直接合成低碳烯烃的过程遵循F-T合成反应机理, 最初是从CO、H2的化学吸附开始:
M+CO→ M-CO(分子吸附)
C-M+CO→ M-CO→ M-C+M-O(解离吸附)
H2+2M→ 2M-H(解离吸附), (M代表金属)
这些活性物种通过不同组合方式进行链增长链支化形成各种烃类化合物和含氧化合物, 也导致不同的F-T反应机理。多年来, 研究者对于这一反应进行了深入研究, 提出了各种反应机理, 主要机理列于表6-2[135]。
| 表6-2 F-T合成机理 |
新碳化物机理提出表面亚甲基生成与本体碳化物无关, 它使碳化物机理被重新认识, 并被认为是解释烃生成的最好机理。然而, 即使在碳化物机理中, 由于活性链C2物种的生成方式不同, 导致碳化物机理出现分歧。从历史上看, F-T合成最初就定性为聚合机理, 而添加CH2结构单元仅能解释烃的生成, CO插入机理被认为出现在含氧化合物的生成过程。至于从CO和H2经表面吸附后如何进行下一步的表面基元反应十分复杂, 研究者考虑有关机理问题有不同层次, 如碳化物机理中表面碳物种生成机理或许与CO插入机理有关等等, 因此对F-T反应机理的研究工作仍需进一步系统考察。
由于目前合成气直接制烯烃的F-T过程受产品烃ASF分布规律的限制, 低碳烯烃很难达到较高选择性, 因此近年来人们开始研究能够突破ASF分布规律限制的合成气直接制低碳烯烃过程的探索, 如开发不同于F-T过程的合成气直接制烯烃过程[136], 该过程采用由甲醇合成组分与甲醇脱水制烯烃组分组成的双功能催化剂, 尽管该过程获得了初步结果, 但要想获得进一步进展, 如何调节各组分功能, 发挥他们的协同效应, 提高烯烃产品的选择性是该过程研究的重点, 高性能催化剂的反应稳定性则是这一过程长期追求的目标。
6.4.4.2 合成气间接转化制低碳烯烃
合成气间接转化制低碳烯烃是指合成气经醇醚等中间化合物合成乙烯、丙烯等低碳烯烃的过程, 该路线的代表过程是合成气基甲醇制烯烃的MTO过程(Methanol to Olefins)。
MTO过程以甲醇为原料, 在一定温度、压力和空速下, 通过特定的催化剂进行脱水反应难过生成低碳烯烃的过程。其主要反应为:
2CH3OH→ C2H4+2H2O
△ G=-115.1 kJ· mol-1,
△ H=-23.1 kJ· mol-1 (6-35)
3CH3OH→ C3H6+3H2O
△ G=-186.9 kJ· mol-1,
△ H=-92.9 kJ· mol-1 (6-36)
4CH3OH→ C4H8+4H2O
△ G=-241.8 kJ· mol-1,
△ H=-150.0 kJ· mol-1 (6-37)
MTO过程是热力学上的放热反应, 低温有利于反应的进行。同时, MTO过程中伴随有低碳烯烃的聚合等二次反应而影响低碳烯烃的选择性, 如何实现产物低碳烯烃的高选择性以及反应热的及时脱除、保持反应系统的稳定性是该过程研究需考虑的重要因素。
以解决MTO过程上述重要因素为目标, 人们分别就MTO催化剂和工艺展开了大量研究。
催化剂是MTO工艺过程的关键技术。由于反应中存在大量水蒸汽, 而且催化剂需在高温下进行, 同时需在较高温度下频繁再生烧炭, 因此催化剂的热稳定性及水热稳定性是影响化学寿命的关键因素。20世纪80年代多采用ZSM-5分子筛及其改性产品, 进入20世纪90年代后则倾向于硅磷酸铝系列分子筛(SAPO), 其中具有强选择性的8元环通道的小孔分子筛SAPO-34备受青睐[137]。在催化剂方面研究较多的机构有美国Mobil公司、德国BASF公司、美国联碳公司、UOP公司和Hydro公司、中国科学院大连化学物理研究所以及中国石化上海石油化工研究院等[137, 138, 139]。
MTO研究的工艺主要包括固定床合成工艺和流化床合成工艺。MTO研究的初期阶段一般采用固定床中试装置, 如德国Karisruhe公司, 装置规模为(20~40) kg· d-1, 反应温度为300 ℃, 压力为(0.11~0.14) MPa, 催化剂是HZSM-5, 甲醇转化率为100%, 乙烯+丙烯选择性为60%。中国科学院大连化学物理研究所的固定床中试装置, 规模为(0.7~1.0) t· d-1, 反应温度(500~550) ℃、压力(0.1~0.15) MPa, 催化剂是P-ZSM-5, 甲醇转化率为100%, 乙烯到丁烯的选择性为86%。为便于反应热的及时移除和催化剂再生, 流化床反应器引起重视, 这种反应器可使反应、再生操作连续化, 大幅度提高MTO的反应效率。UOP/HYDRO-MTO流化床工艺大型示范装置连续运转90多天, 粗工业甲醇的加工能力为0.75 t· d-1, 其中UOPMTO-100催化剂在反应器与再生器之间连续循环操作, 装置运行平稳[139]。中国科学院大连化学物理研究所等采用流化床工艺于2004年进行了甲醇制取低碳烯烃成套工业技术开发(工艺名称DMTO), 建成了世界第一套万吨级(日处理甲醇50 t)甲醇制烯烃工业性试验装置, 并于2006年完成了工业性试验。2010年, 我国利用DMTO技术建设完成了世界首套甲醇制烯烃工业化装置, 装置规模为每年1.8 Mt甲醇生产600 kt烯烃, 该装置一次开车成功并稳定运转[140]。
关于MTO反应机理研究表明, 甲醇转化为烃类的反应包含甲醇转化为二甲醚反应在内的一系列非常复杂反应, 目前已证实甲醇转化为二甲醚的反应, 但第一个C— C键的形成机理仍不清楚。目前被大多数研究者认可的反应机理为:在酸性分子筛催化剂上甲氧基通过与分子筛内预先形成的“ 碳池” 中间物作用, 同时形成乙烯、丙烯、丁烯等烯烃。“ 碳池” 具有芳烃特征, 且反应是平行进行的。通常新鲜催化剂不含芳烃类物质, 而以富含氢和氧的甲醇原料在分子筛微孔内形成芳烃也并非易事, “ 碳池” 一旦形成, 后续形成烯烃的反应则是快速反应(< 0.01s)[141, 142, 143]。
6.5 结论和展望
合成气中枢是非石油路线制取液体燃料和重要化学品的一个重要概括, 合适氢碳比的合成气廉价制备是合成气中枢首要任务, 大力推进廉价合成气制备进而制取化学品, 将实现资源互补最佳利用, 同时减轻环境压力。而围绕合成气中枢任务展开关于合成气化学的基础研究, 将使得合成气中枢在化石能源清洁制取和利用、生物质资源的可再生利用, 特别是非石油路线制取液体燃料和重要化工产品中扮演着愈来愈重要的角色。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|