
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
异丁烷催化脱氢反应机理与失活动力学研究进展
引用本文
王堂博, 王广建, 孙万堂, 牛鑫善, 王芳. 异丁烷催化脱氢反应机理与失活动力学研究进展[J]. 工业催化, 2016,24(4): 1-6.
Wang Tangbo, Wang Guangjian, Sun Wantang, Niu Xinshan, Wang Fang. Advance in catalytic reaction mechanisms and deactivation kinetics of isobutane dehydrogenation[J]. Industrial Catalysis, 2016,24(4): 1-6.
Doi:10.3969/j.issn.1008-1143.2016.04.001Wang Tangbo, Wang Guangjian, Sun Wantang, Niu Xinshan, Wang Fang. Advance in catalytic reaction mechanisms and deactivation kinetics of isobutane dehydrogenation[J]. Industrial Catalysis, 2016,24(4): 1-6.
Permissions
Copyright©2016, 《工业催化》编辑部
《工业催化》编辑部 所有
异丁烷催化脱氢反应机理与失活动力学研究进展
作者简介:王堂博,男,1990年生,在读硕士研究生。
摘要
异丁烯是化工行业重要的基础原料,国内外对异丁烯的需求量逐年递增。仅靠石油催化裂解已无法满足对异丁烯的需求,开展异丁烷脱氢制异丁烯工艺的研究备受关注。综述Cr系异丁烷脱氢催化剂的研究进展,探讨Cr系催化剂的活性中心以及发生在活性中心上的多种反应机理和相应的动力学模型,详述催化剂的失活机理,总结积炭的形成过程,指出Cr系异丁烷催化脱氢反应和失活机理以及相关动力学方面研究的不足,并对未来研究前景进行展望。
关键词:
催化反应工程; 异丁烷; 异丁烯; 脱氢; 动力学; 反应机理
中图分类号:O643.12
文献标志码:A
文章编号:1008-1143(2016)04-0001-06
Advance in catalytic reaction mechanisms and deactivation kinetics of isobutane dehydrogenation
Abstract
Isobutene is an important raw material in petrolchemical industry,and the demand for isobutene is continuously increasing.The technologies for dehydrogenation of isobutane to isobutene have received much attention because the conventional petroleum cracking cannot satisfy the growing demand for isobutene.In this paper,the recent development in chromium-based catalysts for isobutane dehydrogenation was reviewed.The active center of chromium-based catalyst,a variety of reaction mechanisms and their corresponding dynamic model based on the active center were discussed.The catalyst deactivation mechanism and the formation process of carbon deposition were also summaried.The shortage of researches on the reaction and deactivation mechanisms of chromium-based catalysts for isobutane catalytic dehydrogenation and the related reaction kinetic study were pointed out.The future research prospects of the chromium-based catalysts for isobutane dehydrogenation are also outlined.
Keyword:
catalytic reaction engineering; isobutane; isobutene; dehydrogenation; kinetics; reaction mechanism
异丁烯是重要的化工原料, 主要用于制备甲基叔丁基醚、丁基橡胶、聚异丁烯和各种精细化学品[1, 2]。异丁烯下游产品的需求量激增促进了异丁烯产业的飞速发展, 预计2016年异丁烯产量将达到29.78 Mt[3, 4]。我国的异丁烯工业始于20世纪70年代, 虽然近年来我国异丁烯产量有所增加, 但远远不能满足工业发展的需求。异丁烷脱氢转化为异丁烯可缓解异丁烯供不应求的矛盾, 副产物氢气也可用作加氢装置的原料, 是实现天然气、炼厂气资源优化利用的重要途径, 未来应用市场前景广阔。
异丁烷脱氢技术关键在于催化剂的研制与选择, 催化剂决定着异丁烷脱氢工艺的流程和经济效益, Cr系催化剂具有成本低、催化活性好等优点, 但稳定性需要提高[5, 6, 7]。本文对异丁烷脱氢技术中Cr系催化剂的活性中心、反应与失活机理和动力学等方面的研究进行综述。
1 Cr系催化剂的活性中心与反应机理
1.1 催化剂活性中心
异丁烷吸附主要发生在Cr— O位上, 充分研究在异丁烷脱氢环境下Cr— O位的性质对确定催化剂活性中心非常重要。研究表明, 新制备的Cr2O3/Al2O3催化剂中存在大量的表面物种, 包括Cr6+、Cr5+、Cr3+和Cr2+。各个物种的浓度与Cr负载量、催化剂制备过程中的焙烧温度和选择的载体有关。低Cr负载量时, 催化剂含有Cr6+和少量的Cr5+, 此时Cr6+分为可溶性Cr6+和非可溶性Cr6+, Cr6+在载体上的密度随着Cr负载量的增加而增加[8], 最终可溶性的Cr6+稳定在(510)个Cr· nm-2, 非可溶性的Cr6+稳定在2个Cr· nm-2。高Cr负载量时, 除了Cr6+、Cr5+、Cr3+和Cr2+外, 还可以观察到高度对称的Cr2O3和嵌进微晶相中的Cr2
部分研究者认为, 分散在催化剂表面的Cr6+是异丁烷脱氢反应的主要活性中心, Kilicarslan S等[11]通过XPS分析观察到在脱氢反应完成后, Cr6+(2p3/2)在578 eV的峰消失, Cr3+(2p3/2)峰出现。Elzinga E J等[12]认为脱氢反应后, 四面体结构的Cr(Ⅵ )O4转化为八面体结构的Cr(Ⅲ )O6, 八面体的Cr(Ⅲ )O6高度对称, Cr在此结构中没有转移电子的能力, 随着Cr(Ⅲ )O6的积累形成了Cr2O3结晶相, 催化剂失去催化活性。也有学者持不同意见, Weckhuysen B M等[13]通过XANES和紫外可见光谱观察到在反应初期Cr3+由Cr6+形成, 并认为Cr系催化剂脱氢活性主要归咎于Cr3+物种。催化剂表面存在3种类型的Cr3+物种:(1) 由Cr6+反应后得到的氧化还原态的Cr3+; (2) 分散在催化剂表面的配位不饱和Cr3+, 这两类Cr3+物种都具有催化活性; (3) 高度对称的Cr3+和微晶相中的Cr3+, 微晶相中的Cr3+嵌进载体Al2O3中被覆盖, 所以催化活性较低。具有催化活性的Cr3+物种通过吸附气相中的异丁烷分子形成稳定的对称Cr3+物种。
1.2 异丁烷脱氢反应机理和动力学
异丁烷脱氢是气-固多相反应, 动力学模型复杂, 总过程速率由反应最慢的基元反应决定, 即速控步骤。多年来, 由于制备的催化剂物理化学性质不同、反应条件以及测试手段存在差异, 有关异丁烷脱氢反应的机理和速控步骤一直存在争议。在上述活性位研究的基础上, 按照催化剂表面的活性中心数量以及氧离子是否参加反应, 将反应分为单一Cr— O活性位机理、协同Cr— O活性位机理和无氧协同活性位机理。
1.2.1 单一Cr— O活性位机理
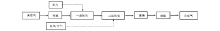
Burwell Jr R L等[14]提出异丁烷脱氢反应是在一个单一Cr— O活性位上发生的, 催化剂上异丁烷脱氢生成异丁烯分为以下3个步骤, 如图1所示。i-C4H9…Cr— O…H表示烷基和氢原子吸附在Cr— O活性位上, a为反应中异丁烷的吸附过程; b为表面反应过程。
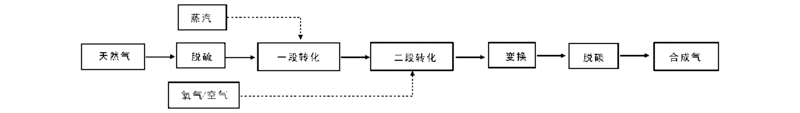 | 图 1 单一Cr— O活性位机理Figure 1 Reaction mechanism of single Cr— O active site |
异丁烷在不饱和的Crn+中心吸附, 然后异丁烷中的C— H键断裂, O— H键和Cr— C键生成, 最后异丁烯脱附, 在催化剂表面生成H2, Crn+活性中心复原[15], 认为反应中以烷烃的吸附过程(步骤1a)作为异丁烷脱氢过程中的速控步骤。
Weckhuysen B M等[16]提出了一个相似的异丁烷脱氢机理, 只是将烷烃吸附与脱附后形成的烷基和氢原子分为两个式子进行表达, 认为该反应机理与Burwell Jr R L提出的机理相似。根据异丁烷脱氢反应机理的结果建立了Cr系脱氢催化剂的动力学模型。
Airaksinen S M K等[17]利用流化床反应器, 在常压和(520580) ℃条件下, 对Cr系异丁烷催化剂的脱氢反应动力学进行研究, 分别以反应过程中异丁烷的吸附和表面反应作为异丁烷脱氢中的速控步骤, 利用Langmuir-Hinshelwood原理建立动力学方程, 采用数学方法优化各参数, 得到动力学方程, 如式(1)所示:
-r1a=
式中, pe、pa和
1.2.2 协同Cr— O活性位机理
Carra S等[18]提出了协同Cr— O活性位机理, 并假设活性位需要Cr和O离子同时存在, 一些步骤发生在同一个Cr— O活性位上, 同时用到Cr和O离子。烷烃在Cr— O键上发生吸附作用, 吸附的烷基与相邻的Cr— O键发生作用, 最后烯烃和氢气从表面脱附, 如图2所示。
 | 图 2 协同Cr— O活性位机理Figure 2 Reaction mechanism of collaborative Cr— O active site |
Carra S以机理中的表面反应(步骤2b)作为速控步骤, 采用反应动力学方程(2)表示:
-r2b=
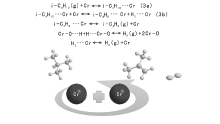
1.2.3 无氧协同活性位机理
Kao J Y等[19]考察异丁烷在无载体Cr催化剂上的脱氢反应, 提出无氧协同活性位机理, 见图3。
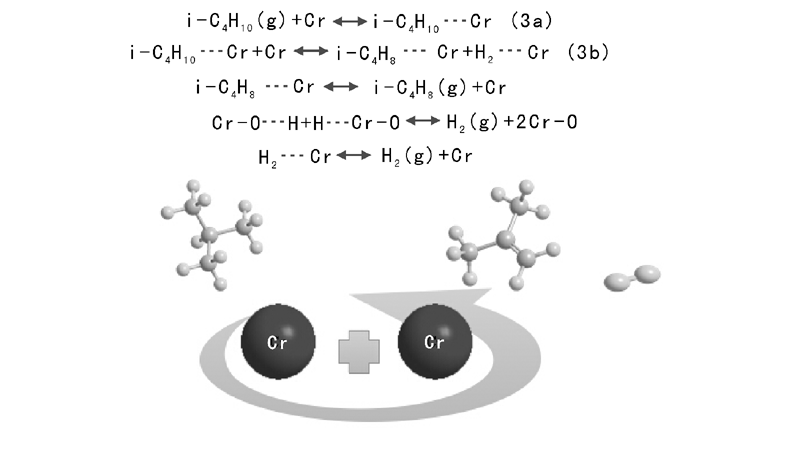 | 图 3 无氧协同活性位机理Figure 3 Reaction mechanism of Cr active site |
Kao J Y以无氧协同活性位机理中的表面反应(步骤3b)为速控步骤, 反应动力学方程见式(3):
-r3b=
Airaksinen S M K等[17]对上述三种动力学模型分别进行了残差平方和SSR分析, 结果显示, 模型(1)的拟合程度最好。单一Cr— O活性位机理中烷烃分子的吸附发生在独立的Cr— O活性位上, 协同Cr— O活性位机理和无氧协同活性位机理中假设的表面反应(速控步骤)则需要多个相同的活性位同时存在。
Airaksinen S M K认为, 单一Cr— O活性位机理更加接近实际烷烃脱氢过程, 所以上述模型的分析结果偏向于单一Cr— O活性位机理。
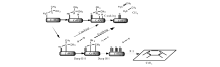
作者认为异丁烷脱氢反应机理可简化由图4表述, 烷烃在催化剂活性位上的吸附过程为反应的速控步骤, 今后的研究可以结合同位素标记法, 对低碳烷烃的脱氢机理进行进一步验证。
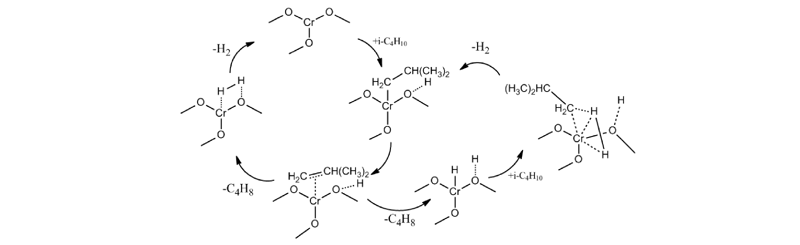 | 图 4 异丁烷在CrOx催化剂上的脱氢反应机理Figure 4 Reaction mechanisms for dehydrogenation of iso-butane on CrOx catalyst |
异丁烷脱氢生成异丁烯的反应机理和反应动力学是异丁烷脱氢反应理论研究的基础, 对异丁烷转化为异丁烯具有重要的指导意义。上述反应动力学方程是利用Langmuir-Hinshelwood原理分析反应机理后得到, 由于未考虑反应中传热、传质等问题, 可能会造成偏差, 导致动力学模型不能客观反映异丁烷脱氢的实际反应历程, 有待于在今后的实验中进一步修正和完善。
2 催化剂的积炭失活机理
异丁烷制备异丁烯的失活过程研究目前主要集中在积炭反应上, 积炭的存在降低了催化剂活性, 并且破坏载体的孔道结构, 使得催化剂进入非永久性失活状态。
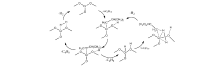
文献[20]通过使用微量振荡天平进行实验, 结果表明, 在反应初期(1040) min, 催化剂的质量随着时间的延长而增加, 随后保持稳定。但是异丁烷转化率随着反应时间的延长依旧下降, 反应形成的积炭使催化剂中毒, 逐渐失去活性。Cr系催化剂表面积炭机理比较复杂, 如图5所示, 积炭通过活性中心深度脱氢、裂化、载体酸性位表面的聚合环化等过程形成。
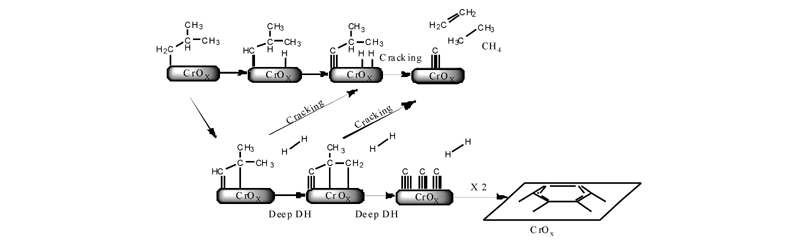 | 图 5 异丁烷脱氢过程中发生的裂化、深度脱氢和积炭反应Figure 5 Proposed process of cracking, deep dehydrogenation and carbon deposition during isobutane dehydrogenation reaction |
Airaksinen S等[21]通过光谱、质谱、色谱的联用对积炭形成过程进行研究, 发现乙酸盐的羧化物和脂肪族烃类在反应初始阶段形成, 然后芳香烃开始在催化剂上积累, 最后催化剂失活。氢气预处理、原料气中加入氧化剂、改善载体酸性位、提高活性组分在催化剂表面的分散度可以有效抑制积炭, 防止催化剂失活。通过分阶段逐步升温的方法进行烧炭可以部分恢复催化剂的活性。但再生过程中烧炭所释放的热量导致活性Cr物种与Al2O3结合, 形成一种稳定的Cr2O3/Al2O3尖晶石结构, 造成催化剂永久性失活。目前异丁烷脱氢制备异丁烯反应失活过程的研究重点主要是积炭造成的非永久性失活。同时, 上述讨论并未涉及碳母体问题, 无法对一些积炭现象做出合理解释, 因此, 有必要研究碳母体的来源问题。
目前, 许多学者提出异丁烷脱氢反应产生的积炭与反应物系的组成和浓度有关。认为催化剂颗粒上的炭分布是由表面向中心减少, 但究竟与何因素相关, 至今仍没有一致结论。Gascó n J等[22]提出传统经典的积炭动力学并沿用至今, 单-多层积炭动力学模型(MMCGM)认为积炭的形成与浓度和温度有关。催化剂表面的积炭浓度与时间关系有两种, 第一种是脱氢瞬间形成的瞬时表面积炭; 第二种是多层积炭。积炭速率由单层瞬时积炭速率和多层积炭速率共同得到, 动力学方程式如下:
-r积炭=
式中, t为反应时间; cc代表积炭总浓度; cm和cM分别代表单层和多层积炭浓度; k1c和k2c分别代表单层和多层动力学常数。
单层积炭量与分散在催化剂表面的活性位数量成正比, 多层积炭伴随着单层积炭的出现而出现, 多层积炭量与被单层积炭覆盖的活性位的量成正比。
3 结 语
异丁烷制备异丁烯过程中的反应与失活机理和反应动力学一直是该领域的焦点与热点, 由于反应过程的复杂性和多样性, 对其反应机理的研究带来巨大的挑战。过去的研究大都集中在本征动力学方面。因此, 需要对Cr系催化剂脱氢反应机理和反应动力学进行更深一步的研究, 尤其加强对宏观动力学方面的研究, 为高性能催化剂的设计制备以及新工艺技术的开发提供理论依据。同时, 研究新型催化剂与新反应体系的动力学机理, 较全面进行相应宏观动力学模型研究是未来研究的方向。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|