



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
第七章 石油化工-精细化工
引用本文
马红, 黄义争, 徐杰. 第七章 石油化工-精细化工[J]. 工业催化, 2016,24(4): 81-113.

Permissions
Copyright©2016, 《工业催化》编辑部
《工业催化》编辑部 所有
第七章 石油化工-精细化工

作者简介:徐杰,男,1958年生,博士,研究员,博士研究生导师,现担任中国科学院大连化学物理研究所-大连洁净能源国家实验室(筹)生物能源部部长、中国化学会催化专业委员会委员、中国化学会均相催化专业委员会委员、中国化学会绿色化学专业委员会委员,任《催化学报》、《Journal of Energy Chemistry》、《Materials Focus》和《Energy and Environment Focus》等期刊编委,大连市第四、第五批优秀专家。长期从事有机催化的应用基础研究,在物理化学、催化化学和有机合成交叉学科的基础上,以具有重要科学意义和应用背景的石油化工、精细化工、能源化学品合成等反应为对象,进行催化新材料、合成新方法、技术新路线等科学研究,为工业应用提供绿色催化合成和资源优化利用新技术和新方法。研究方向主要为烃类催化选择氧化、催化选择加氢、生物质转化和催化新材料制备和应用。近年来,主持国家高技术发展计划(863)项目、国家自然科学基金重点项目、国家“十一五”科技支撑项目、中国科学院知识创新工程重要方向项目、中国科学院东北振兴科技行动计划重点项目等多项,完成多项应用成果和工业化技术,建立烃类催化氧化辽宁省重点实验室。在Angewandte Chemie International Edition、Energy & Environmental Science、Journal of the American Chemical Society和Chemical Communications等期刊发表研究论文200余篇,出版专著1部,申请发明专利200余件,授权80余件,鉴定和验收成果15项,培养毕业博士研究生30余名,多次获得科研奖励。
7.1 前 言
石油化工是工业经济的基础性支柱性产业, 为农业、能源、交通、机械、电子和纺织等多行业提供支撑, 在国民经济中占有举足轻重的地位[1]。2014年, 我国石油和化工行业规模以上企业主营业务收入14.06万亿元, 占全国规模工业主营业务收入的12.8%。基于现代石油化工发展, 精细化工的重要性日益凸显。精细化工产品批量小, 但附加值和生产技术含量高, 其特定功能可满足不同产品需求, 是化工行业中不可或缺的部分[2], 是石油和化工行业最具活力和发展潜力的领域, 近五年行业总产值年均增长率近20%, 目前年总产值超过3.5万亿元。
以石油(包括天然气)为原料生产石油产品和石油化工产品, 高度依赖于石油资源。据统计, 2014年我国原油产量2.11亿吨, 表观消费量5.19亿吨, 进口量3.08亿吨, 进口依存度接近60%。同期我国柴油产量1.76亿吨、汽油产量1.10亿吨、煤油产量0.30亿吨、燃料油产量0.25亿吨, 柴油、汽油、煤油和燃料油总产量约占原油消费量的66%。能源需求的快速增长以及对化石资源消耗速度迅速增加, 使供需矛盾日益突出, 成为制约国民经济可持续发展的关键瓶颈, 如何合理利用有限的石油资源成为重要的战略问题[3]。
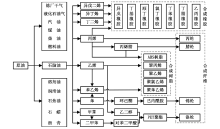
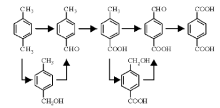
从“ 三苯” (苯、甲苯和二甲苯)和“ 三烯” (乙烯、丙烯和丁二烯)等石油化工关键化学品出发, 经加氢、氧化和氨氧化等重要过程, 可以合成包括对苯二甲酸、乙二醇、苯酚、丙烯腈、2-氯-1, 3-丁二烯等大宗化学品, 用于生产合成纤维、合成塑料和合成橡胶, 即石油化工的“ 三大合成” (图7-1)。
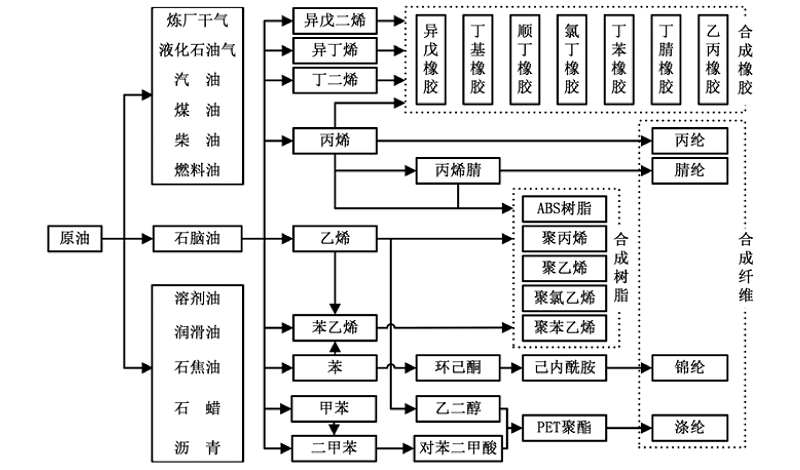 | 图 7-1 石油化工“ 三大合成” 典型路线示意图Figure 1 XRD patterns of the three-way catalysts |
“ 三大合成” 是石油化工中增长最快的产业。2014年, 我国合成纤维产量达4 043.9万吨、塑料制品产量7 387.8万吨(合成树脂产量6 950.7万吨)、合成橡胶产量549.5万吨。如何使“ 三大合成” 原料生产满足市场需求, 是今后需持续关注的焦点[4, 5]。加快石油化工技术开发, 尤其是原始创新技术开发, 突破国外市场和技术垄断, 是满足“ 三大合成” 原料生产和供应的重要技术保障。
催化技术是化工过程的核心技术。采用催化方法生产的化学品占化工产品总量的90%以上, 催化剂产生的价值可达自身价值的5001 000倍。纵览石油化工历经的多次技术变革, 每次技术更新往往伴随着新催化剂及新工艺的出现, 催化剂的革新成为石油化工技术进步与发展的推动力[6]。石油化学工业起始于美国, 20世纪20年代, 美孚石油公司利用炼厂气丙烯催化水合, 制成最早的石油化工产品— — 异丙醇。自此, 石油化工逐渐成为生产化工原料和产品的重要过程。1940年, 合成高辛烷值汽油的催化工艺奠定了现代燃料工业基础。20世纪50年代, 石油化工进入迅速发展时期, 齐格勒-纳塔(Ziegler-Natta)催化剂和乙烯络合氧化催化剂的开发极大促进了石油化工的兴起, 卡尔· 齐格勒(Karl Waldemar Ziegler)和居里奥· 纳塔(Giulio Natta)因相关研究成果获1963年诺贝尔化学奖。20世纪60年代初至70年代末是世界石油化工发展的全盛时期, 相继开发了多种新型催化剂, 成功用于丙烯氨氧化合成丙烯腈、邻二甲苯氧化制邻苯二甲酸酐、合成气制甲醇、甲醇均相羰基化制乙酸等工艺。1973年, 恩斯特· 奥托· 菲舍尔(Ernst Otto Fischer)和杰弗里· 威尔金森(Geoffrey Wilkinson)因对金属有机化合物的研究成果获诺贝尔化学奖, 20世纪80年代, 金属有机化合物茂催化剂使聚烯烃工业得到快速发展。20世纪80年代石油化工进入成熟期, 工厂规模与数量不断扩大, 石油需求量随之增加, 造成石油价格飙高。当时的技术追求经济效益, 大量排污对环境造成巨大压力, 导致石油化工发展速度缓慢。绿色清洁生产的需求和呼声日益突出[8], 国内外相继出台环保政策法规, 制约工厂污染排放, 使工厂不能只注重经济效益。这一改变为环保新催化剂和新工艺进入工业应用提供巨大推动力。例如, 2001年, 威廉· 斯坦迪什· 诺尔斯(William Standish Knowles)、野依良治(Ryoji Noyori)和巴里· 夏普莱斯(Barry Sharpless)因对手性催化加氢和手性催化氧化的研究成果获诺贝尔化学奖, 手性催化剂的应用使反应过程更经济, 同时大大减少毒害废弃物产生, 并可将医药的疗效提高数十倍, 每年产生的直接经济效益达数千亿美元。2005年, 伊夫· 肖万(Yves Chauvin)、罗伯特· 格拉布(Robert Grubbs)和理查德· 施罗克(Richard Shrock)因在金属卡宾催化剂用于烯烃复分解反应方面的成果获诺贝尔化学奖, 该成果可取代大量传统的有机合成方法, 用于设计合成新型药品和先进塑料材料。2010年, 理查德· 赫克(Richard F. Heck)、根岸英一(Ei-ichi Negishi)、铃木章(Akira Suzuki)因在有机合成中钯催化偶联反应的研究获诺贝尔化学奖, 这一成果可使人类制造出复杂的有机分子, 并减少副产品产生, 广泛应用于制药、电子和先进材料等领域。
为实现国民经济可持续发展, 石油化工行业迫切需要解决以下问题:
(1) 资源优化和合理利用。开发可循环再生的资源新路线, 是从根本上解决石油等化石资源短缺问题的有效途径, 但目前该路线在技术和经济上还有障碍, 生产的产品数量和比例仍然较小, 近期难以彻底取代石油、煤与天然气等化石资源。通过技术创新, 实现传统工业改造, 优化利用有限化石资源, 是一条最现实可行的节约化石资源的技术路线, 也是目前技术开发的主要方向。
(2) 知识产权和环境保护。我国石油化工大型装置大多数为引进技术, 投资大, 费用高, 扩产改造受到极大限制和制约。此外, 很多石油化工过程选择性低、资源消耗量大、环境污染严重, 因此, 建立我国自主知识产权的创新技术, 将传统高消耗、高能耗、污染严重的过程升级改造为低成本、高效益的清洁过程, 成为石油化工行业关注的重点。
本章介绍石油化工和精细化工中具有代表性的工业催化过程, 主要介绍通过加氢、氧化、氨氧化、羰基合成和酯化等手段, 从基础有机化工原料出发合成有机化学品, 重点介绍苯加氢制环己烯和环己烷、对二甲苯氧化制对苯二甲酸、环己烷氧化制KA油、苯氧化制苯酚、环己烯氧化制己二酸、丙烯氧化制环氧丙烷等关键过程。
7.2 催化加氢
催化加氢在现代石油化工和精细化工中具有重要地位, 在合成医药、农药等精细化学品中具有广泛应用。加氢反应是有机化合物与氢分子之间发生的反应, 广泛用于不饱和碳氢化合物、含氧及含氮化合物制备碳氢化合物、醇类及胺类产品[10]。通过制备高活性、高选择性以及高稳定性的催化剂进行催化加氢反应, 使得医药、农药等精细化学品工业得到迅速发展, 同时在可再生资源利用以及缓解环境污染等方面也得到重要应用。近年来, 生物质转化快速发展, 尤其是以纤维素(包括半纤维素)、糖和糖醇衍生物以及木质素等为原料, 采用催化加氢裂解反应可以制取醇、芳香化合物等精细化学品[11, 12], 为化学品合成提供了绿色环保、原子经济性好的新途径。
7.2.1 加氢催化剂
金属催化剂是最常用的一类加氢催化剂, 主要包括Fe、Co、Ni、Pd、Pt、Cu、Ag及其合金。除骨架金属外, 常负载在多孔载体上, 并加入适当助剂提高催化性能。金属能够起到解离氢分子、提供活泼氢原子使其在金属表面与有机物发生反应的作用。由于金属催化剂对极性官能团吸附活化较弱, 加氢主要发生在碳碳不饱和双键上。氧化物具有较强极性, 对于底物极性基团的吸附活化有利, 因此, 催化不饱和醛、酮、酸、酯和硝基化合物加氢时, 在催化剂中加入ZnO、Cr2O3等氧化物, 一般可以提高其催化加氢性能。
设计合成高活性、高选择性和高稳定性的金属催化体系, 对于催化加氢制化学品研究具有重要意义。贵金属是常用的加氢催化剂组分, 其中, Pd常用于碳碳不饱和双键加氢, 特别是对于二烯的选择性加氢具有良好的催化活性和选择性。例如, 在除去丁烯中的丁二烯时, Pd可催化丁二烯选择性加氢生成丁烯, 丁二烯不会过度加氢生成丁烷[12]。此外, Pd用于催化卤代有机化合物加氢脱卤反应, 也具有较好活性。
Pt具有加氢、脱氢功能, 广泛用于不饱和碳碳键的加氢反应, 但其活性和选择性一般低于Pd基催化剂。Pt在C=O、C=N的加氢中具有很高的催化活性, 例如, 在α , β -不饱和醛选择性加氢制备α , β -不饱和醇的反应中, Pt更容易催化C=O加氢, 而Pd则倾向C=C双键的加氢。Pt基催化剂还具有在较低温度条件下就能实现良好催化性能的特点。
Ru基催化剂主要用于芳香化合物加氢反应, 对芳环的催化加氢具有高选择性。Ru在水相中催化不饱和有机物的加氢反应具有较高的活性, 例如, 徐杰等合成含有链状钌纳米阵列的PVP-Ru两亲性催化剂, 在水/有机体系中, 对不饱和化合物完全加氢反应具有很高的活性和普适性[13]。Ru对醛、酮化合物加氢也有较好的活性, 例如, 葡萄糖加氢制备山梨醇, 收率接近100%[14]。此外, CO2/CO加氢甲烷化反应中, Ru也被认为是具高催化活性的组分, 而且具有较好的低温活性[15]。
Ni基催化剂是加氢活性高的非贵金属催化剂, 已发展成为一类用途广泛的加氢催化剂, 对不饱和烃或含羰基、硝基、氰基化合物的加氢反应具有催化活性。以Ni为活性金属的催化剂种类较多, 包括骨架Ni、负载型Ni基催化剂等。骨架Ni是具有海绵状孔结构的镍铝合金催化剂, 具有很大表面积, 能吸附大量活化氢, 因此, 骨架Ni催化剂一般具有高的加氢活性和选择性。负载Ni基催化剂是另外一类广泛使用的加氢催化剂, 常用载体有活性炭、SiO2、Al2O3、TiO2、ZrO2等。随着新材料发展, 负载Ni基催化剂的载体拓展到了碳纳米管、碳纤维、介孔碳等材料[16, 17, 18]。
Cu是另一类具有催化加氢活性的非贵金属组分, 具有成本低、比表面积大、活性高等优点, 常用于酯类化合物加氢。例如, 在Cu-Ag/SiO2催化剂作用下, 草酸二甲酯加氢反应转化率在90%以上, 乙醇酸甲酯选择性接近100%[19]。Cu在糠醛加氢反应中也有重要应用, 对糠醛中的羰基加氢具有较好选择性。例如, 使用骨架Cu催化剂, 糠醛接近完全转化, 并高选择性地获得糠醇[20]。此外, Cu基催化剂具有良好的C— O裂解选择性, 例如在丙三醇加氢裂解中, 可提高丙二醇产物的选择性。常用的Cu基加氢催化剂有Cu/SiO2、Cu-Zn催化剂等。夏春谷等使用沉淀-凝胶技术制备了高分散SiO2负载的Cu纳米粒子催化剂, 用于甘油加氢裂解反应, 甘油转化率达到73.4%, 丙二醇选择性达到94.3%[21]。
7.2.2 苯加氢制环己烯
苯催化选择加氢提供了一种以苯为原料制备环己烯的方法。与环己醇脱水、Birch还原等传统方法相比, 该方法的开发和工业应用使环己烯生产成本显著下降, 适合工业化大规模生产[22]。在“ 三大合成” 重要材料尼龙6和尼龙66生产中, 苯选择加氢制环己烯是己内酰胺工艺技术的基本单元之一。与苯加氢制环己烷再氧化制KA油的路线相比, 苯选择加氢制环己烯再水合制环己醇路线耗氢减少1/3, 选择性高, 无酸、酯等副产物, 是一条竞争力强的路线。
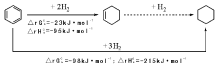
苯催化选择加氢制环己烯的方法包括气相催化加氢和液相催化加氢等。目前研究较多且能应用于工业生产的方法为气液液固催化加氢法。从苯加氢自由能的变化看, 苯加氢反应更容易生成热力学稳定的环己烷(图7-2)。环己烯具有比苯更活泼的非共轭碳碳双键, 很容易一步加氢生成环己烷。气液液固催化加氢法中, 苯选择性加氢合成环己烯是通过使用一个含有苯、环己烯和环己烷的油相以及水、催化剂、气体组成的四相体系, 改变苯、环己烯等物质的传质等动力学条件而进行的。该方法能够在较高转化率下得到较高选择性的环己烯。
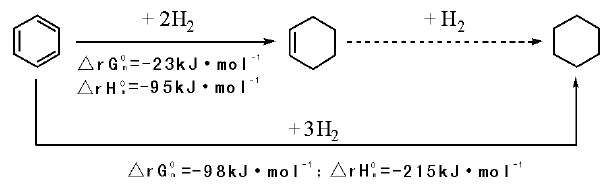 | 图 7-2 苯加氢反应的热力学变化Figure 1 XRD patterns of the three-way catalysts |
1988年, 旭化成公司在日本水岛建成世界上首套苯选择性加氢合成环己烯的工业化装置, 在反应温度(120180) ℃和反应压力(37) MPa条件下, 苯转化率50%60%, 环己烯选择性80%, 副产物环己烷经分离后也可以作为产品出售。旭化成工艺具有反应条件温和、原料消耗低和节能环保等优点。1996年, 我国神马集团尼龙66盐厂引进了该技术, 建成我国首条苯催化选择加氢合成环己烯生产线。随着环己烯下游产品的开发利用, 我国环己烯需求量逐年递增, 国内环己烯产能已不能满足需求。
苯选择性加氢生成环己烯的催化剂需要具备高选择性和高活性。Milone C等[23]发现以三氯化钌为前驱体造成少量氯离子残留, 可提高钌催化剂性能, 这可能是因为氯离子优先吸附在能够强吸附环己烯的活性位上, 并通过与水形成氢键提高催化剂亲水性, 从而利于环己烯的脱附。为了提高环己烯选择性, 一般采用亲水性较好的载体, 包括不溶性硫酸盐及磷酸盐[BaSO4、Zn3(PO4)2]、单金属氧化物(SiO2、Al2O3、ZrO2)及多金属氧化物、分子筛(SBA-15、MCM-41)等。此外, 加入水以及ZnSO4等无机添加剂, 可增加催化剂表面的亲水性, 促使环己烯从催化剂表面及时脱附, 提高环己烯的选择性。在苯选择性加氢催化剂中, 经常加入Fe、Co、Cr、Cu等吸附环己烯能力比Ru强的组分作助剂, 这些助剂可能会从Ru上夺取环己烯, 使环己烯及时脱附, 从而提高环己烯的收率。
7.2.3 硝基化合物加氢制芳香胺
芳香硝基化合物加氢还原制芳香胺, 在精细化工中间体生产中具有重要应用, 可用于农药、 医药、染料和荧光增白剂等合成。
芳香硝基化合物液相催化加氢机理较为复杂, 一般认为其主要历程为硝基依次被还原为亚硝基、羟氨基、氨基。在高纯胺制备过程中, 存在某些中间产物容易生成副产物的问题。例如, 芳基羟胺会导致偶氮或氧化偶氮副产物生成。添加助剂可以提高催化剂的活性与选择性, 例如, 在间二硝基苯催化加氢反应中, 在Ni/SiO2催化剂中添加助剂La2O3, 间二硝基苯转化率接近100%, 间苯二胺选择性为99.5%[24]。La2O3使Ni的晶格常数发生改变, 对Ni表面产生包埋和掺杂作用, 从而削弱NiO与载体SiO2间的相互作用; 另一方面, La2O3在SiO2载体上的单层分散, 也一定程度上削弱NiO与载体SiO2间的相互作用, 有利于Ni物种低温还原, 提高催化剂活性。
卤代芳香族硝基化合物加氢常发生脱卤副反应, 防止其发生是关键。脱卤抑制剂有吗啉、有机胺、三苯基亚磷酸、甲脒盐、噻唑等。水对硝基化合物还原的影响显著, 在溶剂乙醇中加入少量水可以极大促进反应进行。加入30%的水后, Ru催化剂活性提高至原来的4倍[25]。此外, 该方法对于Ni、Cu等其他类型催化剂, 也有显著的促进效应。
Ni、Pd、Pt等单组分贵金属催化剂虽然活性较高, 但选择性差。添加过渡金属组分可提高其催化性能。例如, 添加Fe、Co、Ni、Cu、Cr、Mn等可以提高Pt/TiO2催化对氯硝基苯加氢生成对氯苯胺的活性与选择性, 其中Fe效果最好, 反应速率提高近一倍, 选择性提高至98%[26]。第二组分加入后, 形成了合金, 改变了原来金属的电子性质是其催化性能改善的原因。
7.2.4 脂肪酸及其酯加氢
天然油脂水解得到的脂肪酸中均含有一定量的不饱和双键, 为满足应用需求, 先将双键加氢处理, 转化为饱和或部分不饱和脂肪酸, 可用于生产乳化剂、洗涤剂、润滑油添加剂及油田化学品等产品。天然脂肪酸加氢常采用钒土、硅胶、硅胶钒土或高分子等负载的Ni基催化剂。
脂肪酸及其酯加氢合成脂肪醇是另一重要过程。由于羧酸较酯加氢困难, 一般先将羧酸转化为甲酯, 然后再加氢。目前工业上脂肪酸(酯)的加氢过程主要采用Cu-Cr催化剂, 该催化剂具有催化活性高、反应条件温和的优点, 但Cr对人体的毒害很大, Cr的回收利用以及产品中Cr含量控制难度大是该催化体系的缺点。目前, 无Cr的铜基催化剂研究及应用开发取得了很大进展, 但仍存在反应条件苛刻和能耗高等问题。
脂肪酸及其酯的加氢催化剂主要有铜系和锌系催化剂。铜系催化剂主要有Cu-Cr、Cu-Zn和Cu-Zn-Cr等, 这类催化剂的特点是反应温度相对较低(< 280 ℃), 醇收率高。氢分子在还原态的金属表面发生解离吸附; 同时脂肪酸酯的羰基氧原子被催化剂表面金属活性位吸附, 然后氢原子在催化剂表面发生迁移, 对脂肪酸加氢, 最后, 生成的醇从催化剂表面脱附。锌系催化剂有Zn-Cr, Zn-Al, Zn-Cr-Al等, 这类催化剂活性低于铜系催化剂, 反应温度一般> 300 ℃, 但其对双键不易加氢, 适用于制备不饱和醇。其中, 以脂肪酸酯为原料, 腐蚀性小, 反应活性高。
脂肪酸酯加氢有多种副反应发生, 可显著影响产品质量。烷烃是脂肪酸酯加氢的主要副产物, 是脂肪醇进一步加氢的产物, 也可来源于脂肪醇脱水— — 加氢过程。烷烃的产生与反应条件密切相关, 高温、长时间反应、低压均有利于烷烃生成。脂肪醛不稳定, 易发生变色、生成聚合物等, 严重影响产品质量。醛是加氢中间产物, 醇脱氢也会导致醛的生成, 高温有利于醛的产生, 可通过控制反应条件减少其生成。其次, 副产物高碳醇酯主要来源为脂肪酸酯与生成的高级脂肪醇发生酯交换反应, 或来源于脂肪酸酯水解生成脂肪酸再与脂肪醇发生酯化反应。高碳酯可进一步加氢生成两分子脂肪醇, 该副产物随着反应的进行, 浓度有一极值, 可以通过提高反应温度、控制原料含水量等措施降低该副产物的含量, 也可通过精馏方法将其除去。
7.2.5 生物质来源化合物加氢制化学品
7.2.5.1 生物质多羟基化合物加氢制低碳多元醇
乙二醇、丙二醇等低碳多元醇是重要的化工原料和聚酯单体, 主要用于生产聚酯纤维和防冻剂, 还可用于生产不饱和聚酯树脂、润滑剂、增塑剂、非离子表面活性剂以及炸药等, 用途广泛。2014年, 我国乙二醇表观消费量约1 224.9万吨, 产量约380.5万吨, 进口845.0万吨, 对外依存度接近70%。
低碳多元醇传统生产主要采用石油原料路线。乙二醇主要通过乙烯环氧化制环氧乙烷再水合制得。杜邦公司采用丙烯为原料, 氧化制得丙烯醛, 再水合加氢制得丙二醇; 壳牌公司开发了乙烯环氧化再羰基化、加氢的丙二醇生产工艺。这些路线都需要经过氧化或环氧化等步骤, 技术难度大、效率低、副产物多、物耗高且污染严重。针对上述问题, 近年来开发了煤制乙二醇的煤化工路线, 并进行了生物质资源催化转化制乙二醇、丙二醇等低碳多元醇的技术路线研究。
生物质催化转化制低碳多元醇是一条不依赖化石资源的新技术路线, 采用包括淀粉、纤维素、半纤维素、木质纤维素等作为多元醇的来源, 这些生物质来源丰富, 价格低廉, 是原子经济性高的环境友好过程。随着化石资源的日益减少, 以糖醇为原料生产低碳多元醇越来越受到关注。以碳纤维负载的Ru催化山梨醇氢解, 转化率36%, C2C3醇总选择性79%[27]。由于贵金属价格昂贵, 非贵金属催化剂受到更多关注。负载型Ni基催化剂对山梨醇加氢裂解, 主产物是丙三醇与丙二醇。Banu M等[28]发现Ni-Pt-NaY催化剂可以高选择性地催化山梨醇氢解生成1, 2-丙二醇, 转化率59%, 选择性71%。Cu-ZnO催化剂催化甘油氢解制1, 2-丙二醇, 200 ℃反应16 h, 甘油转化率37%, 1, 2-丙二醇选择性92%[29]。Ni基催化剂也可用于甘油加氢脱氧的过程[30, 31], 催化剂合成条件对其性能影响较大, 采用KBH4处理的催化剂性能最佳。添加Ce可以显著提高Ni基催化剂的活性, 以Ni-Ce/C为催化剂, 在氢压5 MPa和反应温度200 ℃条件下反应5 h, 甘油转化率可达90%, 产物选择性66%, 副产物乙二醇选择性11%[31]。此外, 载体对Ni基催化剂性能影响也较大, Ni/NaX催化剂催化甘油氢解制1, 2-丙二醇性能较好, 甘油转化率为86%时, 产物选择性80%, 副产物主要是乙二醇[32]。ReOx修饰的Ir/SiO2催化剂催化甘油氢解制1, 3-丙二醇, 在反应初始阶段, 1, 3-丙二醇选择性可达67%; 当甘油转化率为81%时, 1, 3-丙二醇选择性降低至38%[33]。
活性炭负载的碳化钨能够将纤维素高效转化为乙二醇, 尤其在少量Ni的促进下, 乙二醇收率高达61%, 为乙二醇的合成提供了一条新途径[34, 35]。中国科学院大连化学物理研究所与企业合作, 开发出葡萄糖-山梨醇转化制低碳醇的催化新材料及其工业应用新技术, 形成具有自主知识产权的非石油路线制乙二醇和丙二醇的新方法, 该技术已在年产20万吨工业装置上得到应用。
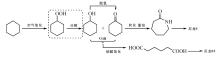
7.2.5.2 木质素加氢制酚类化合物
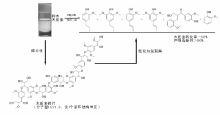
由于石油资源的不可再生性, 生物质可再生资源研究得到广泛关注。木质素是生物质的重要组分, 具有大量的苯环结构, 是制备具有重要应用价值的芳香化合物的潜在可再生资源。木质素加氢裂解制芳香化合物反应中, C— O— C键的选择裂解以及苯环的选择加氢是两大关键问题。徐杰等设计了Ni基催化剂并构建加氢裂解反应环境, 实现了木质素模型化合物、天然木质素以及木质素磺酸盐的选择加氢裂解, 获得优异的反应活性和产物选择性(图7-3)。木质素模型化合物研究是为了模拟木质素的关键结构— — C— O— C键, 通过研究其断键规律, 为天然木质素、废弃木质素等提供研究基础, 在设计的Ni基催化剂上, 模型化合物苯基苯乙醚转化率超过99%, C— O— C裂解选择性达99%以上。在模型化合物研究基础上, 对天然木质素进行了研究。由于天然木质素结构复杂, 含有大量氢键, 造成其难于溶解和降解。针对此问题, 采用合适的醇溶剂体系, 在Ni基催化剂作用下, 桦木木质素催化转化制丙基愈创木酚和丙基紫丁香酚, 转化率50%, 单体酚类化合物选择性达97%。此外, 传统造纸产业上产生木质素磺酸盐等大量废弃木质素, 通过设计高效选择性加氢裂解催化剂, 以乙二醇作介质, 木质素磺酸盐转化率超过60%, 酚类化合物选择性75%95%[36, 37, 38]。
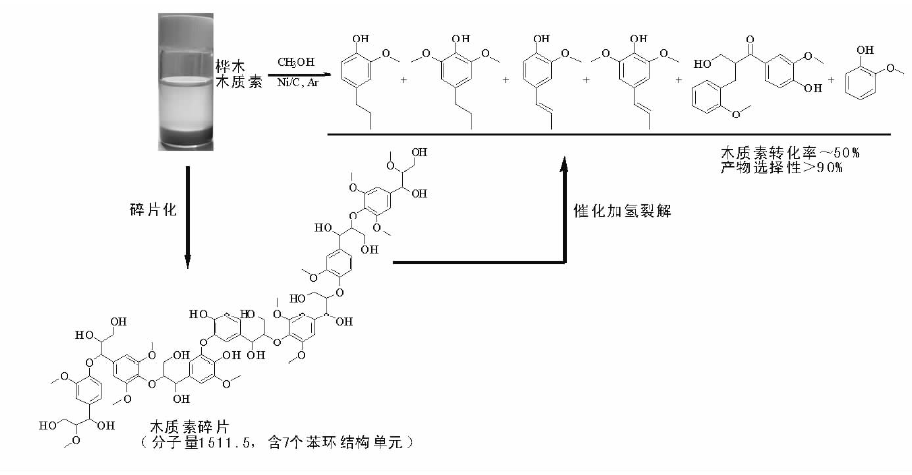 | 图 7-3 木质素加氢路线示意图Figure 1 XRD patterns of the three-way catalysts |
7.3 烃类催化氧化
烃类催化氧化在石油化工中占有极其重要的地位, 是化石资源官能团化高值利用的重要途径, 即利用氧化手段, 使烃分子C— C或C— H键断裂, 生成相应的醇、酮、醛、酸、酯、酚、醚、烷基过氧化物、环氧化物等含氧化合物。据统计, 催化过程生产的有机化学品中, 选择性氧化过程得到的产品约占25%。烃类催化选择性氧化已广泛应用于生产苯甲酸、对苯二甲酸、邻苯二甲酸(酐)、己二酸、马来酸(酐)、反丁烯二酸、丙烯酸(酯)、甲基丙烯酸、环氧乙烷等大宗化学品以及药物、杀虫剂、环氧树脂、染料、香精香料等精细化工产品, 产品用途广、附加值高、需求量大, 在国民经济发展中占据重要地位。例如, 目前我国对苯二甲酸年消费量超过2 700万吨; 环氧乙烷年消费量超过350万吨; 环氧丙烷消费量年超过200万吨; 邻苯二甲酸(酐)、丙烯酸(酯)年消费量都超过100万吨; 己二酸、马来酸(酐)年消费量都超过50万吨。
烃类催化氧化面临巨大挑战。现行工艺存在以下问题:(1) 大多反应效率低。例如, 国内某公司6万吨苯甲酸生产工艺中, 甲苯单程转化率10%15%; (2) 为了维持产物较高的选择性, 有时需要控制转化率在较低水平。在环己烷合成KA油的生产中, 一旦提高反应转化率, KA油的选择性会迅速降低, 为了维持产物较高的选择性(约80%), 环己烷转化率须控制约4%[39]; (3) 烃类催化氧化一般要在较高的温度等较苛刻的条件下实现。过高的反应温度容易造成过度氧化和C— C键断裂, 生成多种、大量副产物, 造成环境污染。几十年来, 各国都投入大量人力物力致力于防止环境污染和解决环境问题, 尤其是近几年加大了对环境污染物的限制力度, 这些传统工艺亟待新工艺替代。
烃类氧化工业过程大多采用分子氧为氧化剂。分子氧为绿色环保的氧化剂, 来源广且价格低廉, 无论从工业应用还是环境保护、资源利用上都十分有吸引力。然而, 在烃类氧化中, 基态分子氧不能与烃分子直接反应。这是由于基态氧分子是惰性的, 处于三线态(3Σ gO2), HOMO中两个单电子自旋方向相同, 不能结合自旋方向相反的成对电子, 而烃分子处于基态时是单线态, 二者自旋禁阻, 很难发生反应。一般情况下, 通过活化分子氧或烃分子这两种途径来实现烃类的氧化。活化氧分子的方式有两种, 一种是激发氧分子到单线态; 另一种方式是利用单电子还原, 依靠还原剂或低价的金属离子转移单电子给氧分子, 使氧分子接受一个电子生成超氧阴离子自由基, 其HOMO中只有一个不成对电子, 表现出自由基活泼的反应性。目前对于分子氧活化的研究主要集中在金属络合物均相催化氧化方面。
烃类活化, 一般指的是活化C— H键。即通过合适的试剂使C— H键的反应活性增加, 从而发生断裂, 变成两个部分。C— H键活化难易程度与其离解能、电子因素、几何因素等有关。键能越强, C— H键越难活化。一般来说, 键能顺序为C— H(sp)> C— H(sp2)> C— H(sp3)(表7-1)。然而, 虽然C— H(sp3)的键能最低, 但却最难活化, 这是电子因素和几何因素共同影响的结果。C— H键的活化方式很多, 譬如过渡金属配合物参与活化, 包括金属中心活化C— H键、配体活化C— H键以及金属配合物先活化其他反应物生成活性物种, 再由这些活性物种活化C— H键[40, 41, 42, 43]。
| 表 7-1 常见烃类C— H键的离解能[44](kJ· mol-1)(%) |
烃分子氧化一般遵循自由基氧化机理。工业上环己烷、二甲苯、异丙苯、异丁烷均有采用自催化氧化法, 在较高温度下直接被空气氧化。为缩短反应诱导期, 一般加入适量自由基引发剂。例如, 液相丁烷氧化制醋酸工业过程, 由(CH3)3COOC(CH3)3引发自由基反应, 分解成叔丁基氧自由基, 随后对烃分子夺氢, 产生烷基自由基。但是大多数烷烃的氧化都包括自氧化过程, 对于这类自由基反应以及包含自由基过程的反应, 无序反应的特征使其难以同时获得高转化率与高选择性。不仅造成资源浪费和环境污染, 而且造成产品分离和纯化困难, 使投资和生产成本大幅度上升。
7.3.1 烃类氧化催化剂
以分子氧为氧源的烃类氧化难度较大。控制氧化反应深度, 提高目的产物选择性一直是烃类选择氧化研究中最具挑战性的难题, 解决选择性难题的途径主要是研制高效催化体系。实践证明, 烷烃的高选择性转化是有可能的。过渡金属催化活化C— H键方面有很多成功的案例, 其中一些反应是在非常温和的条件下进行[45], 尤其是高效仿生催化体系的开发, 获得了十分可喜的成果。另一方面, 高选择性的多相催化剂也不断得以研究, 包括对固体材料的结构设计、表面性质修饰、活性中心的调控以及活性粒子的尺度控制。另外, 新生产工艺的研制开发可以获得高生产率与低成本, 例如, 膜反应器能够控制放热反应, 提高目的产物选择性, 减少污染。
7.3.1.1 金属盐及络合物催化剂
金属羧酸盐和氢溴酸盐是工业常用的氧化催化剂, 常用有钴和锰的乙酸盐、苯甲酸盐或环烷酸盐。例如, 杜邦公司的钴盐法催化环己烷氧化工艺中, 环己烷在可溶性钴盐存在下, 以分子氧作氧化剂, 在(423433) K和0.9 MPa空气压力下, 生成环己醇和环己酮, 然后脱氢生成环己酮, 转化率约4%, 选择性约80%。除环己烷外, 工业上甲苯氧化生产苯甲酸, 也是在623 K下以乙酸钴为催化剂。美国加州大学Mascharak教授合成出一系列烷基过氧化钴配合物[Co(Ⅲ )-OOR][46], 能够催化氧化环己烷、甲苯、环己烯等烃。
近几十年来, 模拟细胞色素P-450单加氧酶研究取得了飞速发展, 最初使用化学计量的单氧原子供体, 如亚碘酰苯(PhIO)、次氯酸钠、过氧化氢、烷基过氧化氢及过硫酸氢钾等作为氧源, 传递单氧原子给中位四芳基卟啉的过渡金属络合物[如5, 10, 15, 20-四苯基卟吩(H2TPP)的铁、钴、锰、锌等络合物], 生成金属卟啉-氧络合物, 之后活泼的高铁氧酰物种氧化烷烃, 实现温和条件下烃类的高选择性催化氧化。以氧分子为氧源时, 要加入消耗的助还原剂(如H2/金属、NaBH4等)来帮助金属卟啉得电子发生还原; 还可以由电化学还原或光辐射辅助催化循环的完成。
研究者对金属卟啉的结构进行了修饰, 一种是在苯基的邻、对、间位引入烷基或卤素取代基, 例如, 2, 4, 6-三甲基苯基卟啉铁(TMPFeCl)苯环上的甲基取代基提供了空间阻碍效应, 避免μ -氧二聚体生成; 而2, 3, 4, 5, 6-五氟四苯基卟啉铁中, 强吸电子F取代基提高了卟啉环的氧化电位, 避免其氧化, 提高了稳定性, 高铁氧酰物种中金属中心的亲电性也提高, 使得缺电子的铁氧酰物种更易于接受电子, 氧化能力增强, 从而提高金属卟啉的活性。例如, 英国约克大学Lindsay Smith实验小组[47]以四-(五氟苯基)卟啉铁Fe(TF5PPCl)作催化剂, 在常压、373 K和无溶剂条件下催化分子氧直接氧化乙苯, 主要产物为α -苯乙醇、苯乙酮和α -苯乙基过氧化氢, 催化剂转化数为2 900。另一种是继续在β 位引入F、Cl、Br等吸电子取代基, 卟啉大环上β 卤基取代的吸电子效应比苯环上卤基取代的效果更强, 使得卟啉环与中心金属离子的氧化还原电位大为提高。例如, 在卟啉苯环邻位引入8个氯原子的TDCPPFeCl比TPPFeCl的氧化电位提高60 mV, 而再在卟啉吡咯β 位引入8个氯原子的(TDCPβ Cl)FeCl比TPPFeCl的氧化电位增加500 mV。从而使得本来必须加入还原剂或者通过光照、电化学辅助手段才能进行的烃类分子氧氧化反应, 在β 卤基取代的金属卟啉催化下成功实现。除卤原子以外, -NO2、-SO3H、-CN等吸电子基进行β 取代也可以使金属卟啉表现出类似的高活性[48]。
随着高活性吸电子取代高卤代金属卟啉的合成, 金属卟啉直接催化分子氧氧化烃类体系(Lyons体系)也发展迅速, 展示出优越的催化特性。20世纪90年代初, Lyons实验小组[49]将一系列多卤代金属卟啉成功用于直接催化分子氧氧化异丁烷成异丁醇, 无需助还原剂、化学计量氧源或光、电化学技术辅助, 转化数超过13 000, 叔丁醇选择性高达92%。
类卟啉金属络合物催化体系, 包括水杨醛席夫碱金属络合物、β -二酮类络合物、冠醚类金属络合物、金属酞菁络合物等, 种类繁多。这些络合物的结构与金属卟啉化合物类似, 在烃类选择氧化上表现出优异特性。β -二酮类络合物用于烃类选择性氧化反应, 可获得高选择性。例如, Gianandrea R等[50]用β -二酮类络合物/异戊醛催化金刚烷氧化, 1-金刚醇选择性高达97%, 但该类催化剂活性较低。冠醚类金属络合物也是一种金属卟啉的重要替代物。这类催化剂应用于分子氧氧化烃分子时, 一般要加入共氧化剂, 例如加入异丁醛, 使之与氧分子形成过酸CH3CH(CH3)COOOH或者烷基过氧化物CH3C(CH3)(CHO)OOH, 转移氧给烃分子。因此, 至少要消耗与底物等物质的量的醛, 并且随之产生的酸需要进一步分离, 并腐蚀设备。
金属卟啉及类金属卟啉催化剂可以使烃氧化反应在较低温度下进行, 产物分布得以有效控制。但是所用配体结构复杂, 合成成本偏高, 配体在反应过程中容易被破坏, 催化剂难以回收再利用。
Gif体系由过渡金属络合物、羧酸、吡啶、还原剂组成, 是美国Barton D H R于1983年开创的一类新的模拟甲烷单加氧酶(MMO)催化氧化体系, 该体系可以在温和条件下催化氧化饱和烃, 最大的特点是氧化碳氢化合物时所表现出独特的区域选择性:仲> 叔> 伯, 2级碳氢键的选择性最高。催化金刚烷氧化时, 金刚烷-2-酮的产率是金刚烷-1-醇的22倍[51]; 催化环己烷氧化时, 转化率高达35%, 环己酮几乎是唯一产物, 没有过度氧化现象[52]。通常情况下, 烃氧化的反应位置一般遵循叔> 仲> 伯由易到难的规律。Barton D H R等研究认为, 反应不经过自由基途径, 其中, 酮不是经过醇继续氧化得到, 而是先由铁离子与吡啶或2-吡啶羧酸络合形成活性络合物, 随后活化氧分子, 进而生成高铁酰活性物种[53, 54]。但该体系的机理存在争议, 有人认为是经过自由基过程, 至今未达成共识。另外, Gif体系存在反应速率相对较慢、使用昂贵且剧毒吡啶溶剂、催化剂易于水解失活等局限, 使得该体系难于进入工业化应用。
7.3.1.2 有机催化体系
有机催化是指用有机物分子作为活性中心来催化有机反应。近年来, 随着现代高等有机合成技术的不断进步与积累, 有机催化体系发展迅速, 有更多手段可用于实现有机分子结构的优化设计以及官能团的变化组合, 进而调变催化性能。
关西大学石井康敬(Ishii)教授发现一种高效催化剂体系(Ishii体系), 由N-羟基邻苯二甲酰亚胺(NHPI)及其衍生物与钴、锰等过渡金属盐组成。该体系普适性强, 能够催化乙烷、异丁烷、金刚烷、环己烷、乙苯等多种烃分子的氧化, 生成相应的醇酮或羧酸。
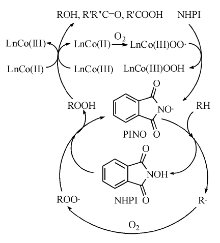
经过单电子转移过程, NHPI生成邻苯二甲酰亚胺-N-氧自由基(PINO), PINO为高亲电性自由基, 可在温和条件下对C— H键夺氢, 实现C— H键的活化(图7-4)。
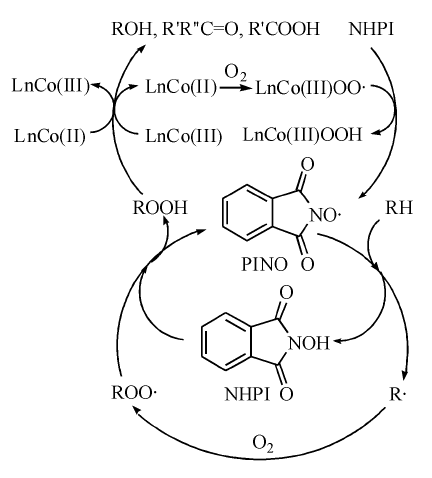 | 图 7-4 Ishii体系催化烃类氧化机理Figure 1 XRD patterns of the three-way catalysts |
利用ESR技术, Ishii Y等[55]检测并证实了反应过程中存在PINO自由基, 在此基础上提出了Ishii体系的催化机理[56]。LnCo(Ⅱ )络合物结合一分子氧形成钴(Ⅲ )氧配合物, 随后对NHPI夺氢生成PINO。PINO对烃分子夺氢, 使其形成烷基自由基, 同时自身回到NHPI。而三价的钴氧配合物能够分解中间产物烷基过氧化氢生成醇酮, 钴离子还原到初始的二价, 形成自身的氧化还原循环。
为了提高NHPI的催化性能, 研究人员对NHPI进行了修饰改进。Wentzel B B等[57]发现, 通过在NHPI苯环上引入吸电子取代基, 可以促进各种取代苯的苄位催化氧化, 相对而言, 在NHPI苯环上引入供电子取代基不利于反应进行。Baucherel X等[58]将NHPI上的羰基C=O变为更具亲电性的磺酰基后, 得到N-羟基邻磺酰基苯甲酰亚胺(NHS), 催化大环环烷烃氧化成环酮的活性高于NHPI。Sawatari N等[59]在NHPI的苯环上连接长碳链醇的酯基, 提高NHPI在烃类化合物中的溶解度, 使得Ishii体系可以在无溶剂条件下催化烃类的氧化, 同时提高催化活性。Einhorn M等[60]对NHPI的苯环进行苯基修饰, 发现3, 4, 5, 6-四苯基-N-羟基邻苯二甲酰亚胺(NHTPPI)不仅溶解性和催化活性均比NHPI高, 而且对应的氮氧自由基也比PINO稳定。除了NHPI衍生物外, N, N', N″-羟基异氰脲酸(THICA)在Co(OAc)2助催化下, 催化分子氧氧化取代甲苯生成取代苯甲酸, 表现出比NHPI更高的催化活性[61]。
除钴盐外, Cu、Cr、Ce、V等多种过渡金属盐被尝试与NHPI组合用于催化烃类氧化反应。例如, Cho S H等[62]报道了Fe/MgO可以提高NHPI/乙醛体系在温和条件下催化苄基C— H键氧化。除这些过渡金属盐之外, 偶氮二异丁腈、烷基过氧化氢、醛类、NO和NO2等也可以协助NHPI完成催化循环。引发剂在反应过程中先分解产生自由基, 再从NHPI上夺氢, 导致PINO的形成, 从而起到催化作用。
利用醌类化合物为电子转移助剂, 与NHPI组成非金属有机催化体系, 是第一个不使用计量还原剂和自由基引发剂, 用于温和条件下烃类氧化反应的有机催化体系。继而利用邻菲罗啉、吖啶黄、甲基紫、甲基蓝等含氮正离子, 在NHPI(或少量NaBr、Br2)存在下, 实现烃类氧化过程中的氧化-还原循环。1, 4-二氨基-2, 3-二氯蒽醌(DACAQ)/NHPI催化芳环侧链亚甲基氧化[63], 具有活性高、选择性好、底物适用范围广的优点。进一步研究醌环上取代基的性质对醌-NHPI体系催化性能的影响, 发现取代基的电子效应和空间位阻效应均有影响。醌环上富电子取代基不利于催化反应进行, 缺电子取代基有利于反应进行, 但并不是拉电子能力越强越好。全卤素取代的对苯醌与NHPI组成的催化体系催化乙苯氧化, 溴取代的苯醌催化效果最好[64]。取代基的个数和体积也在一定程度上影响醌-NHPI体系的催化性能。不同烷基取代的苯醌与NHPI组成的体系在催化乙苯的氧化反应结果表明, 位阻过大、过小均不利于其催化性能的发挥[65]。
7.3.1.3 负载型多相催化剂
多相催化机理与均相催化类似, 金属或金属离子在固体表面的环境与在溶液中类似, 周围的金属或金属离子、氧原子都可以类似认为是特定配体包围在金属或金属离子周围。金属或金属氧化物催化剂能够在较高温度下使用, 这对于需要较高活化能的反应非常有用; 同时固体催化剂使用过程中不存在溶剂或配体的C— H键与烷烃C— H键进行竞争的问题。多相催化剂一般包括以下典型几类:负载型多相催化剂、分子筛、纳米粒子、杂多酸及过渡金属取代杂多酸、均相固载化催化剂。
负载型多相催化剂应用广泛, 已经在不同反应中显示出优良的催化活性与选择性。制备时一般采用浸渍法, 将金属氧化物负载于多孔载体上; 或者通过共沉淀法, 使活性组分进入载体骨架。徐杰等[66, 67, 68]合成多组分金属氧化物负载型催化剂CuMn/C, 在对苯环侧甲基氧化制取代苯甲醛的研究中, 对甲酚几乎定量转化, 醛选择性高达96%, 并应用到邻甲酚催化氧化制水杨醛过程中。
均相催化剂固载在载体后, 兼有均相和多相催化剂的优点。常用的载体主要有硅胶、分子筛和高分子聚合物。固载方法主要有共价嫁接法、离子交换、胶囊化等方法。Das B K等[69]在多孔硅胶上共价嫁接丙酸基团, 得到有机官能团化的多相材料silica-(CH2)2CO2H, 再将羧基与Co(Ⅲ )络合, 所得多相催化剂用于催化乙苯的液相氧化(403 K, O2压力101.325 kPa, 22 h), 表现出优异的催化性能, 转化率达76%, 苯乙酮选择性达94%。但金属络合物多相化后活性往往降低, 限制了其应用。
7.3.1.4 分子筛
分子筛具有高比表面积、规整孔道结构、择形催化性质, 是工业上广泛应用的催化材料。但是分子筛本身不具有或者仅具有弱的氧化还原性质, 用于催化烃类氧化时需要通过离子交换、负载、嫁接及包埋等方法将金属组分作为氧化还原中心引入分子筛。
氧化还原型分子筛磷酸铝(AlPO-n)成功合成以来, 已经有Co、Me、Fe、Cr等多种过渡金属引入磷酸铝分子筛的骨架中, 应用于烃类氧化, 并获得较好进展。钴取代的磷酸铝分子筛中, 催化剂活性与钴的价态有很大关联, 三价钴表现出更好的催化活性[70]。磷酸铝分子筛的孔径大小强烈影响烃类氧化产物的种类。
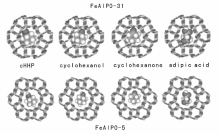
Thomas J M等[71]报道了一系列MeAPO-n分子筛在无溶剂条件下催化空气氧化环己烷, 发现环己烷在0.73 nm的FeAlPO-5上获得65%的己二酸, 而在0.54 nm的FeAlPO-31上却得到77.5%的KA油(图7-5)。推测这是由于环己醇、环己酮在小孔径分子筛中扩散受限, 会进一步氧化生成己二酸, 而链状的己二酸分子易于通过; 在大孔径分子筛中不存在这种限制作用, 因此KA油为主要产物。直链烷烃的氧化, 易发生在— CH2上, 难以发生在端基— CH3上。该课题组[72]使用CoAlPO-18和CoAlPO-34作催化剂, 正己烷可以被氧气氧化为正己醇、正己醛、正己酸和己二酸, 这是非常有价值的突破。徐杰等[73]在磷铝分子筛骨架中同时植入三种金属(FeCoMnAPO-5), 用于催化环己烷氧化, 403 K反应3 h, 转化率达6.8%, 催化活性高于相应单金属组分的磷铝分子筛。
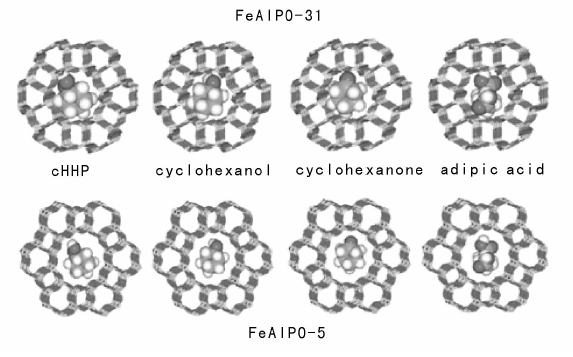 | 图 7-5 环己烷氧化产物环己基过氧化氢、环己醇、环己酮和己二酸在FeAPO-31和FeAPO-5孔道里的相对尺寸Figure 1 XRD patterns of the three-way catalysts |
中孔材料为制备多种高效催化剂提供了可能。中孔材料具有高比表面积(约1 000 m2· g-1)、规则孔结构、强吸附能力, 近年来其催化性能引起了普遍关注。利用SBA-15表面羟基与钴盐反应, 在低负载量时, 可获得高分散的四面体配位Co(Ⅱ )O, Co/SBA-15催化剂在乙苯氧化制苯乙酮中, 表现出高催化性能[74]。Bi-MCM-41用于环己烷氧化, 转化率可达17%[75]。Fe-HMA分子筛作为催化剂, 环己烷转化率可达62.7%; 相同条件下, 其活性比Fe-MCM-41和FeAPO-5高[76]。
7.3.1.5 纳米金属氧化物
多相催化剂中尤为引人注目的是纳米粒子催化剂。纳米粒子通常指尺寸在(1100) nm的微小固体颗粒, 是处在原子簇和宏观物体交界的过渡区域。其表面原子数和体相总原子数之比随着粒径尺寸减小而急剧增加, 显示出明显的体积效应、量子尺寸效应和表面效应。这些特点使其具有高的化学活性, 因此, 纳米粒子催化剂被认为是很具潜力的新型高效催化剂, 应用前景广阔。
对纳米颗粒金属催化剂的研究表明, 该类催化剂具有很高的催化活性。以无定型Co3O4为催化剂, 70 ℃反应(1517) h, 环己烷转化率4.8%; 而在(810) nm Fe2O3催化剂上, 环己烷转化率16.5%, 选择性约90%, 产物中酮醇比为0.4[77, 78, 79]。徐杰等[80]合成纳米Co3O4, 在环己烷氧化反应中, 表现出较高的活性和选择性。平均直径为50 nm的Co3O4表现出最好的催化性能, 120 ℃反应6 h, 环己烷转化率为7.6%, KA油选择性为89.1%。然而,
粒径小的同时也带来另一个问题, 即纳米粒子不稳定, 因此, 保证纳米粒子的尺寸成为研究的关键。但这些催化剂稳定性不足, 例如, 采用纳米Fe2O3为催化剂, 第一次循环使用, 催化剂活性降到新鲜催化剂的70%, 这可能与催化剂的无定型结构、共反应物异丁醛生成酸等因素造成催化剂结构破坏和流失有关。
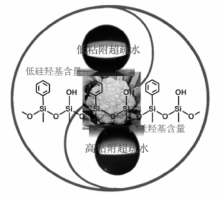
表面疏水修饰是提高金属氧化物在烃类氧化中催化性能的另一个重要手段。徐杰等通过引入亲油性的有机基团, 对多相催化材料表面进行改性修饰, 通过控制表面亲水/疏水基团比例, 可调控材料表面由亲水到疏水和超疏水转变(图7-6), 合成的系列疏水和超疏水型催化剂用于烃类液相选择氧化, 取得较好效果[81, 82, 83]。在乙苯、四氢萘等氧化中, 与亲水性催化剂相比, 超疏水性的纳米氧化硅基过渡金属氧化物显示出更高活性。这些催化剂表面的疏水性能提高, 减弱其对醇、酮等极性产物分子的吸附, 同时增加对烃类分子的吸附能力, 从而提高反应的选择性。
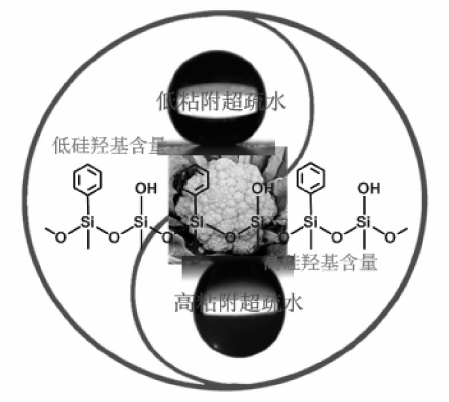 | 图 7-6 超疏水纳米催化材料[82]Figure 1 XRD patterns of the three-way catalysts |
7.3.1.6 杂多酸
杂多酸及过渡金属取代杂多酸是一类含氧桥的多核金属配合物, 具有酸性和氧化还原性, 尤其是在水溶剂或含氧极性溶剂中溶解度大, 形成“ 假液相” 反应体系, 展示出极高的催化活性与选择性。Mizuno N等[84, 85, 86, 87]采用Fe3xNix双金属取代的杂多酸催化氧气氧化环己烷、乙苯、金刚烷等取得较好效果。γ -SiW10{Fe(OH2)2O38}6-在常温、常压下催化分子氧氧化环己烷, 反应96 h, KA油选择性为99%。尽管杂多化合物具有稳定性好等优点, 但直接催化分子氧氧化烃分子的活性不足。
虽然多相催化剂克服了均相催化剂的一些缺陷, 具有很大的应用潜力, 但是多相催化剂也具有一定的缺点需要改进。首先, 催化剂制备处于摸索阶段, 没有明确的理论指导, 而且多相催化氧化的作用原理和反应机理尚不清楚; 其次, 对于烃类氧化而言, 由于产物比原料分子极性大, 活性更高, 不易从催化剂表面脱附, 易产生过度氧化和催化剂团聚, 极大限制了多相催化剂的实际应用, 要实现大规模的工业应用还有一定距离, 需要进一步深入研究。
7.3.2 环己烷氧化制环己酮和己二酸
7.3.2.1 环己烷氧化制环己酮
环己烷催化氧化制环己酮, 是重要的石油化工中间过程。环己酮是合成尼龙6、尼龙66等聚酰胺类纤维和工程塑料的重要原料, 在医药、农药、油漆、涂料、染料、橡胶、油墨、塑料等行业应用广泛。据统计, 2013年, 全球环己酮总产能约738.7万吨, 产量517.9万吨; 我国产能200.0万吨, 产量130.3万吨, 表观消费量129.0万吨。
工业生产环己醇和环己酮主要有无催化氧化法、硼酸酯氧化法和钴盐催化氧化法。目前, 最成熟的环己烷氧化制环己酮工业生产技术是荷兰矿业公司(DSM)开发的无催化氧化技术, 世界上90%以上的环己酮经过该路线生产。无催化氧化法又称过氧化物氧化法, 该技术第一阶段为环己烷无催化氧化生成环己基过氧化氢; 第二阶段在NaOH等大量碱存在下, 使环己基过氧化氢分解为环己醇和环己酮混合物, 转化率3.5%4.2%, 总选择性76%81%。此法面临以下主要问题:(1) 转化率低。转化率和选择性之间存在很大矛盾, 当转化率超过3.5%, 转化率每提高1%, 选择性下降4%以上; (2) 选择性低。氧化过程中, 有19%24%的环己烷被转化为副产物而浪费掉; (3) 污染大。在分解过程中, 加入的NaOH等大量碱转化为碱渣, 每生产1吨环己酮, 产生碱渣(150180) kg, 此外, 还有大量废水产生。
硼酸法是以硼酸或硼酸酐为催化剂, 使环己烷先氧化生成硼酸环己醇酯, 然后水解成环己醇。该工艺的弱点在于:工业运行中回收硼酸非常复杂, 经济成本高, 增加公用工程消耗, 并且硼酸很容易造成设备严重堵塞, 装置不能长期稳定运行。
钴盐法操作简单、技术成熟。在该工艺中, 环己烷在可溶性钴盐存在下, 以分子氧为氧源, 在温度(150160) ℃, 0.9 MPa空气压力下, 生成环己醇和环己酮, 环己醇再脱氢生成环己酮, 转化率约4%, 选择性约80%。由于转化率低, 大量未反应的环己烷不得不在分离后重新回到反应原料中, 造成能耗高、设备利用率低等问题。并且由于选择性差, 生成大量难以处理的副产物, 造成环境污染、资源浪费严重。1992年, 日本Sumitomo公司开发出异辛酸钴催化剂, 转化率提高到5.9%, 但选择性降至71.7%。1997年, 日本Ube公司通过添加N-甲基咪唑催化剂, 使选择性提高到82.9%, 但转化率下降到3.9%。
国内环己酮和己二酸生产装置几乎全部引进国外技术, 例如, 中国石化巴陵石化、南京东方公司等的环己酮生产装置引进荷兰矿业公司(SDM)技术, 中国石油辽阳化纤的己二酸装置引进法国技术, 河南神马的己二酸生产装置引进日本旭化成技术, 中国石化石家庄化纤公司的苯甲酸生产氧化装置引进意大利米兰实验室(SNIA)技术。因为没有自主知识产权, 引进费用高, 扩产改造困难, 成为制约行业发展的主要瓶颈。
综上所述, 目前环己酮的工业生产存在原料转化率低、目标产物选择性差、反应条件苛刻以及对环境造成污染等, 必须解决以下关键问题:(1) 节约资源和技术升级。环己烷氧化过程是资源消耗量大的石油化工过程, 污染严重, 必须对该传统石油化工过程进行技术升级; (2) 扩产改造和知识产权问题。目前国内环己酮大型生产装置为引进国外技术, 没有完整知识产权, 扩产改造费用高, 应用和发展受到限制。克服这些问题的关键在于自主产权的高效催化剂开发。
7.3.2.2 环己烷氧化制己二酸
己二酸是环己烷氧化的另一重要产物, 是生产尼龙66的单体, 也可用于生产造纸化学品、医药、润滑剂等化学品。2012年, 全球己二酸产能349万吨, 消费量285万吨; 我国产能115万吨, 消费量77万吨。环己烷氧化制己二酸工业生产是基于20世纪40年代初美国杜邦公司开发的两步合成工艺[88]。该工艺采用钴盐为催化剂, 是最早实现工业化的环己烷氧化法, 被称为环己烷氧化的经典方法(图7-7)。首先由环己烷合成KA油, 然后由硝酸氧化KA油为己二酸。但是两步合成法不仅产率低, 而且产生N2O气体等副产物。
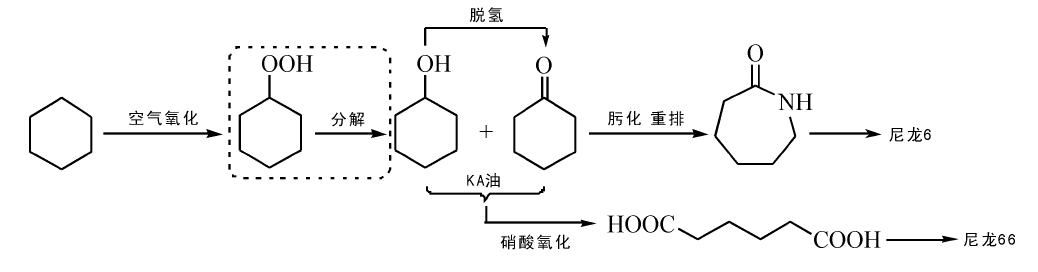 | 图 7-7 环己烷催化氧化制KA油、己内酰胺和己二酸Figure 1 XRD patterns of the three-way catalysts |
7.3.3 对二甲苯氧化制对苯二甲酸
对二甲苯氧化制对苯二甲酸是重要的石油化工过程。对苯二甲酸是氧化产品中需求量最大的石油化工产品, 也是聚酯、树脂和化纤行业最重要的单体, 广泛用于生产涤纶纤维、聚酯薄膜、聚酯瓶。
如图7-8所示, 原油经过炼制得到石脑油, 然后经重整等过程制得对二甲苯, 对二甲苯经过氧化、结晶、分离、干燥等过程, 生产出精对苯二甲酸。在以上产业链中, 对苯二甲酸是原油的产品, 同时又是化纤的原料。2014年, 我国对苯二甲酸产能近4 500万吨, 表观消费量2 744万吨, 产量2 656万吨, 连续多年保持世界最大对苯二甲酸生产国地位。
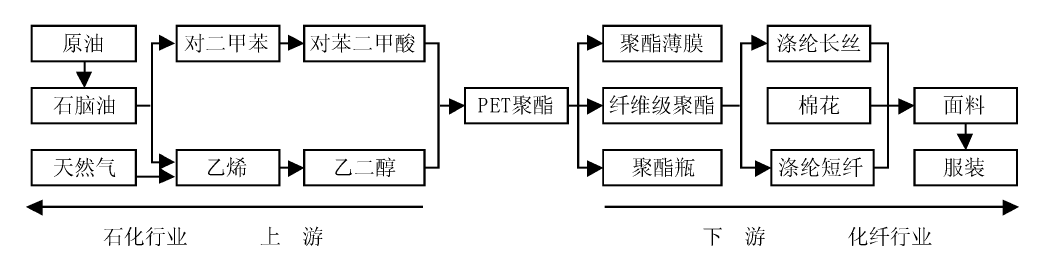 | 图 7-8 石油化工-PET聚酯-化纤产业链示意图Figure 1 XRD patterns of the three-way catalysts |
对二甲苯氧化制对苯二甲酸的主要生产工艺有BP-Amoco工艺、ICI工艺和三井工艺, 包括液相氧化、结晶、离心、干燥、加氢、精制、分离等步骤; 其中, 对二甲苯的液相氧化是核心单元。尽管不同工艺精制和分离路线不同, 但氧化过程均采用Co-Mn-Br催化剂、以空气为氧化剂、醋酸为溶剂条件下进行。BP-Amoco工艺技术是目前生产对苯二甲酸的最成熟工艺, 催化效率高, 醋酸溶剂消耗少, 产品质量好, 反应在(160240) ℃、(12) MPa条件下, 以空气为氧源连续进行, 装置中对二甲苯进料浓度约20%。氧化产物经钯碳催化剂加氢精制, 除去少量醛类副产物, 得到精对苯二甲酸。
国内对苯二甲酸生产技术和工艺基本以进口技术为主, 存在较大技术风险。最近几年虽有多套对苯二甲酸装置上马, 但市场变化复杂, 外国少数公司从上游原料、生产技术、关键设备等控制着中国对苯二甲酸行业。因此, 开发自主知识产权的对苯二甲酸催化剂及生产工艺, 对于提高企业的核心竞争力, 打破外国公司的垄断, 真正让中国的聚酯行业做大、做强具有重要意义。
对二甲苯氧化是一自由基连续反应, 对二甲苯侧链的两个甲基依次被氧化为苄醇基、醛基和羧基, 如图7-9所示。主要中间产物有对甲基苯甲醇、对甲基苯甲醛、对甲基苯甲酸、对羟甲基苯甲酸、对醛基苯甲酸等。
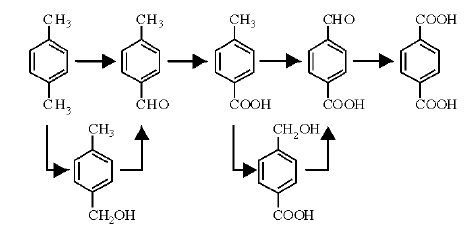 | 图 7-9 对二甲苯氧化制对苯二甲酸路径Figure 1 XRD patterns of the three-way catalysts |
苯环上甲基的氧化类似于典型烷烃的金属盐催化液相氧化, 催化循环机理如图7-10所示。
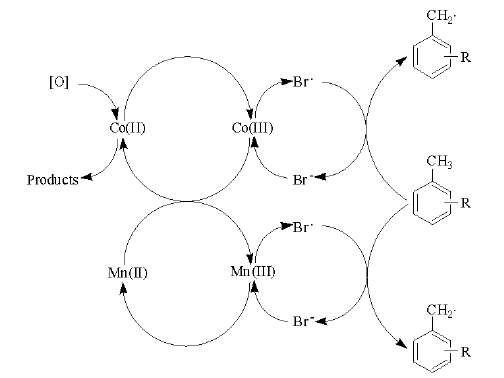 | 图 7-10 Co-Mn-Br催化循环机理Figure 1 XRD patterns of the three-way catalysts |
目前, 对苯二甲酸生产过程中, 溴在提高催化氧化反应活性和降低反应温度等方面具有不可替代的作用。由于采用大量含溴催化剂, 反应过程中排放大量的溴甲烷和其他含溴化合物。以现有对苯二甲酸生产规模计算, 我国对苯二甲酸生产过程中溴化物用量每年超过65 000吨。大量溴的使用和排放, 对环境造成极大持久性污染。此外, 溴对金属反应器、管线和阀门等组件的腐蚀非常严重, 不仅增加装置建设费用, 而且容易发生管道泄露、堵塞等非正常停车, 造成运行不稳定, 并存在安全隐患。
长期以来, 改进氧化催化剂及反应工艺, 提高反应效率, 减少对二甲苯氧化制对苯二甲酸催化剂的用量及成本, 减少副产物生成, 提高产品质量等是国内外关注的核心问题, 也是实现对苯二甲酸清洁生产的重大需求。例如, 三井-Amco工艺采用鼓泡塔反应器, 通过提高催化剂中Co与Mn比和溶剂比, 可降低反应温度和压力, 并减少副产物生成; 美国Praxair技术公司开发以富氧为基础的对二甲苯氧化工艺, 通过采用纯氧或富氧空气, 尽可能提高液相中的氧气浓度, 使进料对二甲苯迅速反应消耗而使液体中烃浓度尽可能降低, 提高反应效率; 在Co-Mn-Br催化体系中添加Hf、Zr等金属组分可提高催化剂活性, 促使对二甲苯氧化反应程度加深[89, 90]; 通过添加三氟乙酸等强酸或少量碱金属、碱土金属也可以促进反应效率提高; 通过改变反应介质, 例如, 在超临界二氧化碳或水体系中反应, 也有助于减轻原有工艺的腐蚀问题, 提高产物选择性。上述研究在提高反应效率、减少Co用量等方面取得一定进展, 但催化剂体系中仍需使用大量的溴。中国科学院大连化学物理研究所开发的对二甲苯氧化制对苯二甲酸关键新技术, 研制出具有自主知识产权的高效催化剂体系, 提高氧化反应活性, 在反应效率和对二甲苯消耗不变情况下, 可大幅降低溴的用量。与企业合作, 该技术完成了年产10万吨规模的应用试验, 连续开车运行结果表明, 该技术对于降低消耗和减轻溴化物污染排放, 具有显著效果。
7.3.4 甲苯和乙苯氧化
甲苯、乙苯等烷基芳烃的选择氧化是具有代表性的烃类氧化过程, 是有机合成过程中重要的反应类型之一。苯甲酸是甲苯氧化的重要产品, 主要用于生产苯甲酸钠、苯甲酸钾、苯甲酸酯增塑剂、醇酸树脂和饲料添加剂。2014年, 全球苯甲酸产能约73.5万吨, 我国总产能49.2万吨。甲苯的另一个重要氧化产品苯甲醛是医药、染料、香料和树脂工业的重要原料, 主要用于生产月桂醛、月桂酸、品绿等, 还可用作溶剂、增塑剂和低温润滑剂等。
甲苯氧化制苯甲酸是意大利SNIA技术生产己内酰胺的主生产单元之一, 工业生产过程采用钴、锰等金属的可溶性盐为催化剂, 在液相条件下选择性氧化甲苯。通过控制适宜的操作条件, 控制甲苯转化率, 提高苯甲酸选择性。例如, 采用钴盐为催化剂, 在(160180) ℃进行甲苯本体氧化, 甲苯单程转化率约15%, 苯甲酸选择性约85%, 副产物苯甲醛选择性约为5%[91]。添加溴化物可以显著提高甲苯液相氧化的转化率及苯甲醛的选择性, 但产品中会有溴代产物生成。
由于苯甲醇、苯甲醛等中间产物比甲苯更容易氧化, 因此难以在甲苯高转化率下获得苯甲醛的高选择性。在反应初始阶段, 甲苯转化率低, 苯甲醛选择性高于50%, 但反应液中苯甲醛浓度累积到一定值, 会有苯甲酸生成, 苯甲醛选择性迅速降低[92]。甲苯选择性氧化制苯甲醛反应为强放热反应, 可自发进行, 且低温对反应有利[93]。
近年来, 多相催化分子氧氧化甲苯合成苯甲醛受到广泛关注。该方法选择性高, 且产品不含卤素。该方法按照反应条件不同, 可以分为气相反应和液相反应。在气相条件下, 以V-Ag-O为催化剂, 在反应温度300 ℃和空速8.9 L· (g· h)-1条件下, 甲苯转化率为9%, 苯甲醛选择性为61%, 副产物苯甲酸选择性为31%[94]。在液相条件下, 采用CuFeOx/
Al2O3等多相催化剂, 也可获得较高的苯甲醛选择性。目前这两种方法的原料转化率均较低, 需要进一步提高。
乙苯氧化产物苯乙酮属于精细化工产品, 可用于制香皂和香烟, 也可用于制纤维素醚、纤维素酯和树脂等的溶剂以及塑料的增塑剂, 还可用于合成医药中间体。目前, 苯乙酮的工业生产主要有两种, 一种是乙酐与苯的Friedel-Crafts反应生产工艺, 在计量的无水三氯化铝催化下, 乙酐与苯发生酰化反应生产苯乙酮, 该方法具有产率高、产品纯度好等优势, 缺点是伴生大量的酸性废水; 另一种是异丁苯法生产甲乙酮, 联产苯酚, 副产苯乙酮的工业生产路线。每生产1吨甲乙酮可联产(1.261.28)吨苯酚, 副产(0.20.26)吨苯乙酮, 丁烯利用率67%70%。异丁苯法的特点是反应条件温和、设备腐蚀较轻, 但工艺过程复杂, 苯乙酮产量受制于甲乙酮的市场销量。乙苯催化氧化直接合成苯乙酮无疑是一条绿色经济的生产路线。
7.3.5 苯氧化制苯酚
苯酚是重要的有机化工原料, 主要用于生产酚醛树脂和双酚A、己内酰胺、烷基酚等产品, 并用作染料、医药、炸药、农药等精细化学品的原料(图7-11)。自1923年采用苯磺化法实现苯酚的工业化生产以来, 苯酚生产发展很快。据统计, 2014年全球苯酚产能为1 321.2万吨; 我国产能197.5万吨, 表观消费量165万吨。
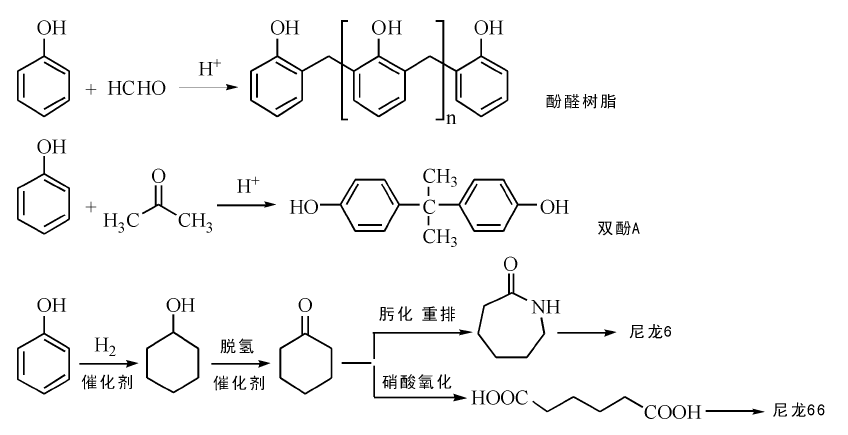 | 图 7-11 苯酚主要下游产品Figure 1 XRD patterns of the three-way catalysts |
最早的苯酚化学合成工艺路线是磺化法, 此后相继发展了氯苯法、环己烷法、甲苯-苯甲酸法、联产丙酮的异丙苯法等, 目前苯酚生产主要采用异丙苯法(图7-12)。异丙苯法生产苯酚技术成熟, 经济性较好。据统计, 世界上90%以上的苯酚通过此工艺生产。该方法是以苯和丙烯为原料, 烷基化反应生成中间产物异丙苯, 然后再经空气或氧气氧化生成过氧化氢异丙基苯, 最后, 过氧化氢异丙基苯经过酸或树脂处理, 得到产物苯酚, 联产丙酮。异丙苯法生产苯酚经过三步反应, 流程长, 生产成本受副产物丙酮价格影响, 生产过程中有大量含酚废水排出, 环境污染严重。
 | 图 7-12 异丙苯法生产苯酚路线图Figure 1 XRD patterns of the three-way catalysts |
苯直接氧化法制苯酚是最具挑战的路线。苯在温和条件下极难被氧化, 在高温下虽然容易发生反应, 但也容易发生深度氧化, 变成焦油状物质或者完全氧化成CO2和H2O。因此, 直接氧化法制苯酚的关键是研究开发具有高活性、高选择性的催化剂, 同时还要选择合适的氧化剂。直接氧化法的氧化剂主要包括N2O、过氧化氢、氧气/空气等。N2O来源较少, 价格最高; 过氧化氢居中; 氧气/空气来源最广, 价格最低。
7.3.5.1 过氧化氢氧化法
苯氧化制苯酚的典型催化体系为芬顿试剂, 由Fe2+与过氧化氢组成。催化原理是在Fe2+作用下, 过氧化氢生成羟基自由基, 羟基自由基很容易加成到苯环上发生羟基化反应。该方法苯酚转化率较高, 但产物与催化剂不易分离。Karakhanov E A等[95]合成了水溶性含铁相转移催化剂, 可将苯转移到水相, 过氧化氢对其氧化得到苯酚, 然后转移到有机相中。
自从具有独特的拓扑结构的钛硅分子筛催化剂(TS-1)合成以来, 在有机化合物选择氧化中的催化作用得到广泛研究。在苯与过氧化氢选择氧化制苯酚的反应中, TS-1表现出较好的催化活性和很高的选择性。在温度323 K下, 苯转化率可达到68%, 苯酚选择性96%, 过氧化氢利用率为70%[96]。中孔结构V-MCM-41分子筛及V-MCM-48分子筛对苯液相氧化制苯酚也表现出一定活性[97], 实验结果表明, 增加催化剂酸性, 活性有所提高。
Niwa S等[98]报道了苯一步合成苯酚的膜法催化工艺, 523 K下, 苯转化率为2%16%, 苯酚选择性为80%97%。该工艺的转化率与选择性还有待改进。
由于过氧化氢价格较昂贵, 苯酚生产的原料成本是对该路线工业化提出的最大挑战。但是随着过氧化氢新生产技术发展以及原位生成过氧化氢技术的出现, 成本有望逐渐降低, 这条路线有望成为合成苯酚的新工艺。
7.3.5.2 N2O氧化法
1983年, Ren T等[99]使用N2O为氧化剂, 苯一步氧化成苯酚, 在温度550 ℃下, 以V2O5/SiO2为催化剂, 苯转化率10%, 苯酚选择性70%, 但催化剂易失活和结焦。对于该反应的机理, 认为位于ZSM-5孔道内的α -Fe为活性中心, 在α -Fe作用下, N2O分解产生活性物种α -O, 然后与苯反应生产苯酚。Panov等发现含铁的酸性ZSM-5分子筛在(500900) ℃水蒸汽处理后, 可极大提高苯酚产率, 苯酚选择性接近100%, 催化剂的稳定性也得到提高。通过吡啶原位吸附表征认为, 铝从骨架脱除, 形成Lewis酸中心, 同时Brö nsted酸中心也转化为Lewis酸中心, 提高了催化剂活性。并且经高温水蒸汽处理, 不会破坏沸石的分子筛结构。但处理时间过长, 可能引起活性中心胶结, 导致活性反而下降。Waclaw A等[100]发现, 质子酸中心的数量与强度和催化剂的失活密切相关, 高硅铝比的Fe/ZSM-5表现出好的活性与稳定性; 当晶粒直径较大时对反应不利, 由于受到扩散阻力, 产物苯酚易吸附在H+、Na+等阳离子位上, 从而形成积炭导致催化剂失活; 焙烧温度(700900) ℃, 有利于形成活性中心α -Fe, 并降低Brö nsted酸中心的数量, 活性最好。
文献[101]报道了具有等级孔结构的Fe/ZSM-5分子筛催化N2O对苯羟基化反应的活性, 发现与传统的Fe/ZSM-5分子筛相比, 催化剂的寿命、活性与选择性都得到提高, 其中反应活性提高至原来的4倍。这可能是因为多级结构分子筛有利于分子的快速扩散。由于采用该方法的苯酚装置受到相联结的已二酸生产装置规模的牵制, 只适用于较小规模的苯酚装置。
7.3.5.3 氧气/空气氧化法
从经济成本的角度讲, 氧气/空气是最便宜的氧化剂, 而且来源方便, 不会产生环境污染等问题。以氧气/空气为氧化剂直接氧化苯制苯酚是最有开发价值和应用前景的途径, 也是国内外研究开发的主要趋势。采用的催化剂主要是以过渡金属为活性组分的催化剂, 但该方法苯酚收率都很低。目前, 氧气/空气氧化苯制苯酚的研究方法主要有气相法和液相法。
气相法是高温反应, 反应过程存在催化剂易失活、有副产物生成、产物易深度氧化等问题。Yamanaka I等[102]以V4Mo8Ox/SiO2和V8Mo4Ox/SiO2为催化剂, 在温度833 K和水蒸汽压力大于10 kPa条件下, 苯酚收率为4.3%, 选择性为40%; 水蒸汽的存在抑制了苯的完全氧化。以Cu/ZSM-5为催化剂, 在温度673 K和N2存在下, 苯酚最高收率为1.6%, 副产物有少量的CO和CO2, 而高硅铝比的ZSM-5载体可以提高苯酚选择性[103]。
Passoni L C等[104]采用Pd(OAc)2-杂多酸催化剂体系, 用V(HOAC)∶ V(H2O)=1∶ 2的溶液作为溶剂, 氧气为氧源, 在温度403 K下反应4 h, 苯转化率为15%, 苯酚选择性为70%, 但催化剂回收后使用选择性差。Liu Y Y等[105]以杂多酸[(C4H9)4N]5[PW11CuO39(H2O)]为催化剂, 在温度323 K下反应12 h, 苯转化率达到9.2%, 苯酚选择性为91.8%, 但反应体系需要加入共还原剂抗坏血酸。研究发现四价钒是苯与氧气合成苯酚的活性中心, 四价钒通过“ reductive activation” 方式还原活化分子氧, 产生活性氧进攻苯环, 得到产物苯酚[106]。
尽管氧气/空气氧化苯制苯酚原子经济性好, 符合绿色化学发展趋势, 但是苯酚收率较低, 尚不能满足工业化应用要求。
7.3.6 环己烯氧化制己二酸
目前工业上采用环己酮氧化法生产己二酸, 即环己烷先氧化成环己酮和环己醇(KA油), 然后将KA油通过硝酸氧化制得己二酸。硝酸氧化过程产生的N2O造成严重的环境污染。
有研究者致力于环己烷一步催化直接氧化制己二酸, 催化剂主要为过渡金属。该方法的主要难点在于产品纯度达不到生产聚酯的要求, 提纯成本高, 限制了其工业应用, 如何提高选择性、降低分离提纯成本是今后研究的重点。BASF和ARCO公司开发了1, 3-丁二烯催化加氢羰基甲氧基化制备工艺和氧化羰基化路线, 这两种方法不用苯作为原料, 原子利用率高, 避免了N2O的产生, 但该法操作条件高, 仍需要改进。
以环己烯为原料, 采用过氧化氢为氧化剂, 直接氧化合成己二酸是一条绿色合成路线。反应路径如图7-13所示。该方法原子经济性及环己烯利用率高。但由于过氧化氢价格高, 利用率还有待提高, 该方法目前成本高于传统路线, 提高过氧化氢利用率, 降低生产成本是该方法实现工业应用的关键。
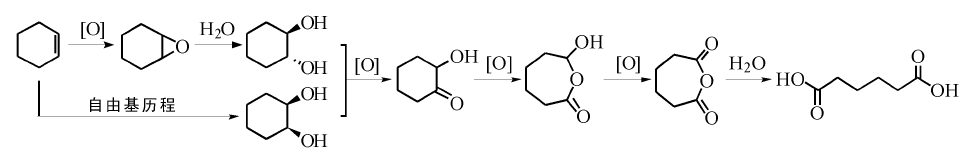 | 图 7-13 环己烯氧化制己二酸可能路径Figure 1 XRD patterns of the three-way catalysts |
7.3.7 丙烯环氧化制环氧丙烷
环氧丙烷是重要的基础有机化工原料, 主要用于生产聚醚、不饱和树脂、丙二醇、非离子表面活性剂、油田破乳剂、农药乳化剂等产品, 广泛用于汽车、建筑、食品、烟草、医药及化妆品等行业。2014年, 全球环氧丙烷产能1 026.5万吨, 消费量850.0万吨, 我国产能284.5万吨, 产量208.4万吨。
环氧丙烷传统生产方法是氯醇法, 每吨产品消耗(1.41.5)吨氯气, 副产3.5吨氯化钙, 并产生40吨废水, 污染严重。共氧化法克服了氯醇法的腐蚀大、污染严重等缺点。异丁烷共氧化法是将异丁烷和丙烯进行共氧化, 生产环氧丙烷, 副产异丁烯。乙苯共氧化法是将乙苯与丙烯共氧化, 生产环氧丙烷, 副产苯乙烯。共氧化法缺点在于工艺流程长、装置设备投资大以及对丙烯纯度要求较高。
过氧化氢催化丙烯环氧化生产环氧丙烷是一种新生产技术, 反应产物为环氧丙烷和水, 无副产品产生, 是一种环境友好的清洁生产方法。2003年, 陶氏化学和巴斯夫开始合作开发过氧化氢法(HPPO)生产环氧丙烷, 采用原位生产的过氧化氢作为氧化剂。中国科学院大连化学物理研究所开发了反应控制相转移催化体系, 用于双氧水直接氧化丙烯制环氧丙烷, 为环氧丙烷提供了环境友好的合成途径。
由于过氧化氢的制备成本比较高, 近年来, 人们试图利用氢气和氧气原位生成过氧化氢氧化丙烯。该过程的催化剂主要为Pd、Ag、Au和Pt等的负载型催化剂。研究发现最好的催化体系是Au负载在含Ti载体上, 包括金红石相TiO2、Ti-MCM-41、Ti-β 、TS-1、Ti-TUD等[107]。该方法选择性高, 但丙烯转化率和过氧化氢利用率需要进一步提高。
从理论上讲, 空气是最理想的氧化剂。但是对于丙烯和氧气的反应, 完全氧化反应的速率非常大, 导致环氧丙烷选择性非常低。研究该反应的机理、寻找合适的催化剂抑制完全氧化反应、提高产物的选择性是该方法需要解决的难题。
7.4 醇催化氧化制醛(酮)
醇氧化制醛(酮)的过程广泛应用在香精、医药及食品添加剂等行业。传统的醇氧化方法多采用化学计量氧化剂, 有活性二氧化锰、Sarrett试剂、Jones试剂、Dess-Martin试剂等, 具有活性高、选择性好的优点, 适用于实验室小规模合成。计量氧化法成本高, 产生大量废弃物, 易造成环境污染。分子氧催化醇氧化是公认的绿色过程, 采用的催化剂主要有Pd、Ru、Pt等贵金属配位化合物、Cu、V等廉价金属催化剂以及TEMPO等有机催化体系。
7.4.1 贵金属催化体系
Pd、Ru、Au、Pt等贵金属配位化合物在醇氧化中表现出独特的催化性能。1977年, Schwartz J等将PdCl2/NaOAc均相催化体系用于脂肪醇氧化[108], 该体系使用氧气为氧源, 能氧化脂肪仲醇生成相应的酮。20世纪90年代后期, 均相Pd催化醇氧化有了显著进展, 研究发现有机配体和溶剂强烈影响Pd的催化性能。研究者通过使用不同含氮配体, 开发了高效Pd均相催化体系, 实现常温常压下醇羟基的选择性氧化[109]。Pd活性对溶剂非常敏感, Stahl S S发现Pd(OAc)2在吡啶或DMSO中具有高催化性能, 认为DMSO、吡啶能够通过与Pd0络合配位作用阻止Pd0团聚(图7-14)[110, 111, 112, 113]。
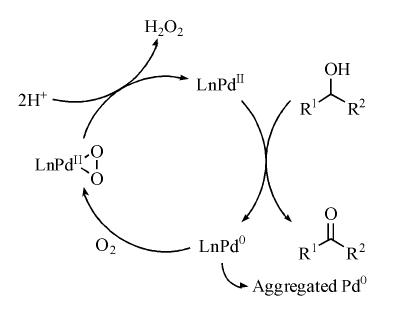 | 图 7-14 Pd(OAc)2/DMSO体系催化醇氧化机理Figure 1 XRD patterns of the three-way catalysts |
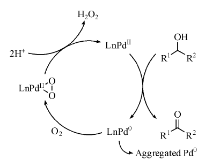
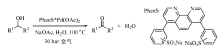
通常需要加入一定量碱促进钯配位化合物的催化效果, 但钯易发生聚集形成钯黑, 严重影响钯的催化效率。Sheldon R A小组使用PhenS阻止钯的聚集, 合成了水溶性有机络合物PhenS* Pd(OAc)2催化剂(图7-15), 较高转化率实现水溶性好的脂肪仲醇和环醇水相氧化为醛, 但苯甲醇转化率不高, 这可能与其在水中的溶解度相对较低有关。研究认为Pd(Ⅱ )催化剂的高活性和稳定性依赖于Pd0的快速氧化, 避免其团聚失活[114]。
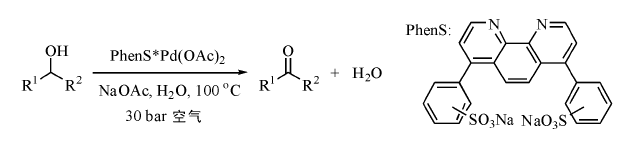 | 图 7-15 PhenS* Pd(OAc)2催化醇氧化反应Figure 1 XRD patterns of the three-way catalysts |
一般情况下, 由于Pd与C=C有强配位作用, Pd催化烯丙醇类氧化反应效果不理想, 而Pd(Ⅱ )-Hydrotalcite催化剂正好克服了这一缺点, Uemura小组[115]在水滑石[Mg6Al2(OH)16CO3· 4H2O]表面引入Pd(OAc)2/pyridine配合物, 该催化剂对于烯丙醇类的氧化反应表现出很好的催化效果。
Ru用于催化氧气氧化醇的早期研究中, 主要采用RuCl3催化剂。随后研究者发现与均相Pd催化体系类似, 配体对Ru络合物的催化性能影响很大。如[RuCl2(p-cymene)]2/Cs2CO3能够高效地氧化脂肪醇和芳香醇[116]。除RuCl3和Ru(PPh3)3Cl2等单核钌外, 双核钌和三核钌配合物也用于脂肪醇催化氧化成醛反应, 表现出高的催化活性。多相Ru催化剂也表现出高催化活性。Kaneda等开发的羟磷灰石负载Ru催化剂(Ru/HAP), 可以高效催化醇氧化, 底物适用范围广, 循环使用3次后仍有较高活性, 但Ru负载量相对较大[117]。Ru/γ -Al2O3能够在80 ℃和常压下氧化苄醇、含硫含氮的杂环芳香醇、含双键的烯丙醇、脂肪伯醇、仲醇等, 而且不需要碱等添加剂, 催化剂能够循环使用。该催化剂使伯醇容易氧化成酸, 加入少量氢醌可以抑制酸的生成[118]。在八面沸石分子筛孔道内合成的纳米RuO2催化剂, 能够催化氧化多种类型的醇, 并表现出高的活性与选择性。例如, RuO2/FAU能实现苯甲醇氧化的化学计量转化, 循环使用5次未见明显的活性降低, 相同条件下普通RuO2仅获得16%的收率[119]。
与其他贵金属催化剂不同, 目前研究的Au催化醇氧化主要是多相体系。研究发现金催化剂表现出明显的尺寸效应, 例如, Tsunoyama H等[120]将金纳米粒子负载到PVP上, 应用于醇的氧化, 反应在300 K水相中进行, 可以将芳香醇选择性氧化成芳醛, 但对于脂肪醇效果不好。动力学实验结果表明, 1.3 nm的金粒子活性大大高出9.5 nm的金粒子, 这从另一方面说明O2在催化剂上的吸附是影响催化效果的关键因素。
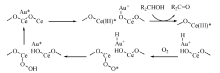
载体自身的性质, 包括氧空位、碱性、尺寸等都能对金纳米粒子产生强烈影响。负载在纳米二氧化铈上的纳米金是一种性能独特的催化剂, Au/CeO2不仅能高选择性氧化醇生成相应的羰基化合物, 而且在无溶剂和常压条件下活性也非常高。作者认为, 在二氧化铈上的活性氧空位作用下产生的带正电荷的金, 可能参与了氧化历程(图7-16)[121]。
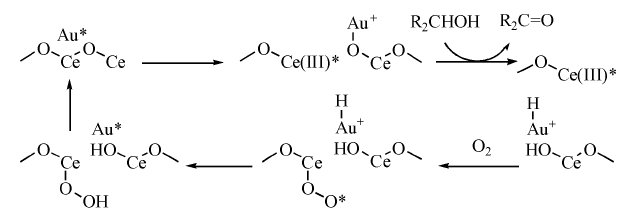 | 图 7-16 Au/CeO2催化氧化醇的反应历程Figure 1 XRD patterns of the three-way catalysts |
如何获取稳定性高的小尺寸纳米金粒子是关键。改变载体, 调控金的催化性能是一条重要途径。含苯基的聚合物负载金催化醇氧化, 实现了室温条件下氧气氧化醇合成醛(酮)的过程, 聚合物链上的苯环稳定了金纳米颗粒, 降低了金与底物醇的配位能力, 使该催化剂具有较宽的底物普适性[122]。利用无机硅铝酸盐分子筛HY超笼, 对平均粒径为1 nm的金纳米簇进行空间限域(图7-17), 提高金纳米簇的稳定性和催化活性, 在丙三醇和5-羟甲基糠醛的催化氧化反应中, 获得高催化性能[123]。
 | 图 7-17 HY超笼限制的纳米Au簇催化剂Figure 1 XRD patterns of the three-way catalysts |
大量使用碱是金催化醇氧化面临的另一大问题。在反应体系中加入NaOH等碱助剂, 可以加快羟基脱氢反应速率, 避免羧酸产物在催化剂表面吸附和毒化。曹勇等将金纳米粒子负载在Ga-Al复合氧化物上, 在80 ℃和常压下, 以甲苯为溶剂, 能够实现多种醇的选择氧化。在无溶剂、无碱性添加剂条件下, 160 ℃可高活性催化氧化α -苯乙醇, 转化频率(TOF)达到25 000h-1[124]。
Pt用于水相醇氧化具有优异的催化活性, 通常以Pt/C、Pt/Al2O3等负载型催化剂用于醇羟基的催化氧化。采用5%的Pt与1%的Bi负载在活性碳上, 以水为溶剂、空气为氧化剂, 高效实现了仲醇氧化生成酮[125]。使用苯甲醇还原负载在PS-PEG amino resin上的Pt盐, 合成了平均直径在5.9 nm的Pt纳米粒子, 实现了水相氧化苄醇、烯丙醇、脂环醇、脂肪醇, 且能实现催化剂的循环使用[126]。虽然贵金属催化醇氧化效果非常好, 但由于价格昂贵, 在一定程度上制约了大规模应用。
7.4.2 非贵金属体系
非贵金属催化具有来源方便、价格低廉的优势, 在醇氧化反应中逐渐受到重视。近年来发现多种高效的非贵金属催化体系, 其中关于铜的研究比较多。铜的催化最初起源于对半乳糖氧化酶的模拟, 这种酶能够高效、高选择性催化苄醇类及烯丙醇类的有氧氧化, 并且在催化氧化过程中有过氧化氢生成。研究人员模拟半乳糖氧化酶中铜与氮、氧的配位结构, 合成了若干个配位化合物, 其催化醇的氧化性能相似, 氧化过程中也有过氧化氢生成。
邻菲啰啉(phen)被用于铜的配体, 与CuCl、肼二甲酸叔丁酯(DBADH2)组成复合催化体系, 在二倍化学计量的K2CO3存在下, 实现醇的氧化。该体系具有较宽的底物普适性, 但对脂肪伯醇的氧化效果不理想[127]。随后发现使用氟苯代替甲苯作为溶剂, 可以用催化量的K2CO3或t-BuOK代替二倍化学计量的K2CO3, 不过该体系对脂肪伯醇的氧化效果仍然不好[128]。后来研究发现, 当向体系中加入催化量的N-甲基咪唑, 可以实现Cu基催化体系对脂肪伯醇的高效选择性氧化[129]。作者认为, Cu(phen)与脂肪醇生成的醇盐比较稳定, 不能进一步反应, 而导致催化剂在氧化脂肪伯醇时失活, 当加入N-甲基咪唑, 通过其在催化剂活性位上竞争配位, 可以阻止催化剂的失活。
铜在醇氧化中还具有催化脱氢能力。铜不仅可以催化醇高温脱氢, 在温和条件下催化脱氢方面也取得了一些进展。Zaccheria F等[130]将铜负载到三氧化二铝上, 以醛或酮为氢受体, 催化氧化多种醇都取得了很高的转化率和选择性, 该催化剂具有非常好的稳定性和循环性。钒基催化剂用于氧分子氧化醇的反应, 近年来也受到关注。徐杰小组报道了VOSO4/NaNO2催化体系催化醇的选择氧化, 发现该体系对芳香醇的选择性氧化非常有效, 苯环上取代基的电子效应对氧化效率无明显影响, 反应历程涉及VⅣ /VⅤ 的氧化-还原循环。该体系的优点是不怕水, 少量水还能够缩短反应诱导期, 促进反应进行[131]。该小组还报道了Cu(NO3)2/VOSO4催化氧气氧化5-羟甲基糠醛, 室温反应48 h, 原料定量转化为2, 5-二醛基呋喃(图7-18)[132, 133]。
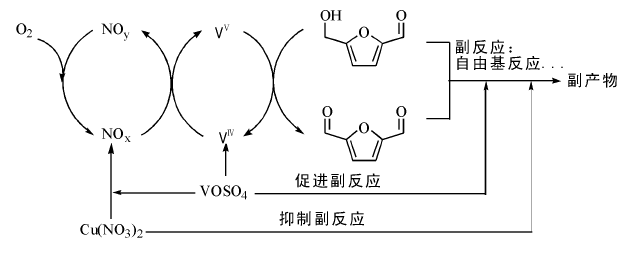 | 图 7-18 Cu(NO3)2/VOSO4催化5-羟甲基糠醛氧化制2, 5-二醛基呋喃[132]Figure 1 XRD patterns of the three-way catalysts |
非贵金属固体催化剂的研究日益受到重视, 不过这方面取得的进展不及均相催化。Suib等报道了锰氧化物八面体分子筛H-K-OMS-2催化以空气为氧化剂的醇选择氧化, 该催化剂用于氧化芳香醇和烯丙醇的收率都很高, 且可重复使用[134]。Ni-Al类水滑石催化剂能够用于烯丙基醇、芳香醇和α -酮醇的氧化, 在氧化α -苯乙醇时循环使用6次仍能保持高的催化活性[135]。Wang F等研究了Mo-V-O氧化物在醇氧化反应中的活性, 发现该催化剂可以氧化伯醇生成醛, 且苯环上取代基对氧化效率有明显的影响, 而仲醇则发生脱水反应生成烯烃[136, 137, 138]。
7.4.3 有机催化体系
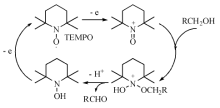
近年来有机催化剂在醇氧化反应中的应用发展迅速, 其中最具代表性的有机催化剂是2, 2, 6, 6-四甲基哌啶-N-氧自由基(TEMPO), 这是一种具有分解过氧化氢、捕获自由基、猝灭激发态等多重作用的高稳定性自由基。在醇的选择性氧化中, 醛酮的过度氧化一般是自由基反应, 利用TEMPO的特点, 使醇的氧化停留在醛酮, 避免过度氧化进行。TEMPO催化醇氧化的机理如图7-19所示。
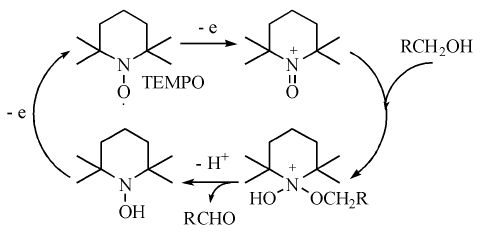 | 图 7-19 TEMPO催化醇氧化的反应机理Figure 1 XRD patterns of the three-way catalysts |
早在1965年TEMPO就用于醇氧化, Golubev报道了计量的氮氧正离子作为氧化剂, 将伯醇氧化成醛; 后来发现用4-甲氧基TEMPO生成的氮氧正离子可以氧化伯醇、仲醇及二醇。TEMPO用于催化则是以间氯过氧苯甲酸、过氧硫酸盐、高碘酸(H5IO6)以及次氯酸钠等计量氧化剂为最终氢受体, 这些氧化剂可将TEMPO氧化成TEMPO+, 然后TEMPO+再氧化醇成相应的羰基化合物。
最初, TEMPO作为催化剂应用于醇氧化, 是以NaClO为氧化剂。1984年, Semmelhack M F等[139]报道了以分子氧为氧化剂, CuCl/TEMPO催化醇到醛(酮)的氧化, 转化频率(TOF)916 h-1。随后开发了以过渡金属为助催化剂的催化体系, 实现了醇的高效选择性氧化。代表性的体系如:Dijksman A等[140]报道Ru(PPh3)3Cl3/TEMPO可以催化氧化活泼醇和非活泼醇到相应的醛或酮; Bjorsvik H R等[141]发现Mn(NO3)2、Co(NO3)2和Cu(NO3)2作为助催化剂, 可以氧化伯醇和仲醇为醛、酮。Buffin B P等[142]以醋酸钯与水溶性联喹啉衍生物的配合物为催化剂, 水相分子氧选择氧化醇类化合物, 加入少量NaOAc时, 该催化体系在仲醇氧化到酮中获得较高收率。徐杰等报道了VOSO4/TEMPO催化氧化醇羟基的新体系, 该体系以分子氧为氧源, 能够在较温和条件下氧化苄醇、脂肪仲醇等[143]。固体催化剂易于回收使用, 采用固体金属氧化物作为助催化剂也有报道。徐杰等研究发现共沉淀法制备的CuMn复合氧化物, 可以促进分子氧为氧化剂的TEMPO催化醇氧化[144], 以VOPO4/TEMPO为催化剂、分子氧为氧源, 在水溶液中实现了3, 4-二甲氧基-苯甲醇的选择性氧化[145]。
使用过渡金属作助催化剂, 可能会带来污染问题。最近报道的不含过渡金属的助催化剂体系, 开辟了TEMPO催化氧分子选择性氧化醇的新领域。Liu R H等[146]以Br2-NaNO2为助催化剂, 实现了TEMPO催化醇氧化反应, 反应机理如图7-20所示。该小组使用亚硝酸叔丁酯和HBr分别替代NaNO2和Br2, 改良了催化体系, 提高了催化效率[147]。Wang X L等[148]发现HCl-NaNO2为助催化剂, 也能高效实现醇的选择性氧化。
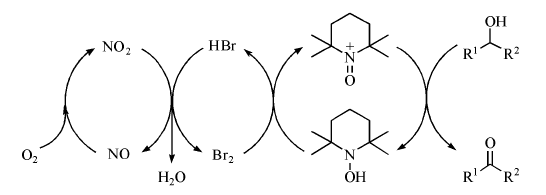 | 图 7-20 NaNO2/Br2/TEMPO催化醇氧化反应机理Figure 1 XRD patterns of the three-way catalysts |
采用合成简单、低廉的有机催化剂替代TEMPO也逐渐受到重视。Lahtinen P等[149]采用9, 10-二氨基-菲催化3, 4-二甲氧基-苯甲醇氧化合成醛, 100 ℃反应3 h, 原料转化率可达80%, 加入助催化剂FeCl3或CuCl2, 转化率可达99%。采用N-羟基邻苯二甲酰亚胺替代TEMPO, 以VO(acac)2为助催化剂, 选择氧化仲醇合成酮, 而伯醇氧化则生成了羧酸[150]。不含过渡金属的催化体系2, 3-二氯-5, 6-二氰基-1, 4-苯醌(DDQ)/NaNO2/NHPI也可催化芳香醇、脂肪醇生成相应的醛(酮)[151]。徐杰等采用H2SO4-NaNO2产生的NOx为催化剂, 实现了以O2为氧化剂醇氧化合成醛的过程[152]。使用更易操作的固体磺酸和亚硝酸异戊酯分别替代H2SO4和NaNO2, 高效地实现了苯甲醇的选择性氧化[153]。
7.5 氨氧化
氨氧化也称氨解氧化, 即通过C— H键的氮官能团化, 在氨存在下, 使烃催化氧化生成相应的腈。目前已经开发的烃类氨氧化工业过程, 主要是通过复合氧化物催化剂实现。在烃类催化转化中, 丙烯或丙烷氨氧化制备丙烯腈是最重要的反应。丙烯腈是三大合成材料— — 合成纤维、合成橡胶、合成塑料的基础原料。2014年, 全球丙烯腈产能673.2万吨, 消费量540.0万吨; 我国产能151.8万吨, 产量131.4万吨, 表观消费量183.2万吨, 腈纶、ABS/SAN、丙烯酰胺对丙烯腈的消费量分别约占45%、31%和16%, 聚醚多元醇、包括丁腈橡胶在内的其他方面约占4%[154]。
甲基芳烃氨氧化可以得到芳香腈, 包括苯甲腈、邻苯二腈、间苯二腈、对苯二腈以及烟碱甲腈等重要化学中间体。烟碱甲腈可以水解为烟碱甲酰胺或烟碱酸, 用于维生素B合成。邻苯二腈是合成酞菁染料的中间体。间苯二腈经氯化反应可制得四氯间苯二腈, 用于农药杀菌剂和防霉剂, 具有高效、广谱、低毒、低残留等优点。
除了已经实现工业应用的气相氨氧化过程, 最近报道了液相氨氧化[155], 将甲基芳烃氨氧化为腈, 进一步水合为酰胺。液相氧化为传统的高能耗氨氧化转化过程, 提供了一条更为绿色的解决思路。
反应温度控制对产物选择性具有重要影响。如丙烯氨氧化生成丙烯腈的反应热为-515 kJ· mol-1, 反应会生成氢氰酸、乙腈, 另外, 碳氧化物生成以及氨气燃烧为氮气是放热反应, 使总放热提高到(530660) kJ· mol-1, 进一步增大了控制反应温度的难度, 为此, 反应通常在流化床装置进行[156]。
催化剂是氨氧化反应的关键。随着研究者对影响反应活性关键因素认识的深入, 催化剂得以不断改善。经过40年发展, 丙烯氨氧化制丙烯腈的催化剂使产物收率增长了近一倍, 达到80%以上, 催化剂中含有20多种元素, 每一种元素对于优化催化效果都起到关键作用。随着丙烯价格上涨, 从更为廉价的丙烷出发制备丙烯腈变得日益重要, 这对催化剂提出了新的要求。
7.5.1 丙烯氨氧化
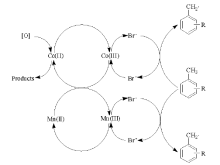
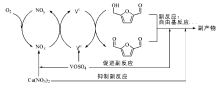
用于丙烯氨氧化的钼酸铋工业催化剂由多组分的Bi-Fe-Ni-Co钼酸盐构成, 并含有Cr、Mg、Rb、K、Cs、P、B、Ce、Sb和Mn等添加剂。这些复合氧化物分散在硅胶上, 用于流化床反应装置。经过40多年研究开发, 丙烯氨氧化制丙烯腈的催化剂已发展到第5代。目前, 钼铋铁催化剂占主导地位, 约90%的丙烯腈工业装置使用的是钼铋铁催化剂。催化剂中三价的Bi/Fe/Cr钼酸盐提供催化剂活性中心, 二价的Ni/Co/Fe/Mg钼酸盐主要作用是增大催化剂的氧化再生速率。催化剂中需要过量的Mo来实现最佳的催化性能:过量的Mo不仅可以将不同钼酸盐桥联起来, 还可以补充因redox循环而损失的Mo[Mo以MoO(OH)2的形式流失]。丙烯中— CH3上的氢被催化剂中的氧脱除, 形成π -烯丙基配合物是整个反应的速控步骤[157, 158, 159]。随后在没有氨时, 氧插入形成丙烯醛; 氨存在时, 氨通过与氧离子交换形成NH2— , 插入烯丙基中间体生成丙烯腈。催化剂中消耗的氧随后被氧气补充[160, 161]。
选择性丙烯氨氧化催化剂表面的活性位点具有三种重要功能, 分别对应于催化剂中特定的金属组分[162]:Bi3+、Sb3+或Te4+作为脱除丙烯中— CH3上氢的组分; Mo6+或Sb5+作为烯烃化学吸附以及氧或氮插入组分; Fe2+/Fe3+或Ce3+/Ce4+作为氧化还原电对, 增强晶格氧在催化剂体相到表面之间的迁移能力。
7.5.2 丙烷氨氧化
目前生产丙烯腈的工艺中, 丙烯成本约占生产总成本的67%[163]。相对丙烯而言, 丙烷更为廉价, 具有很强的竞争力。早在1961年, 已有研究者从热力学上推断了以丙烷为原料生产丙烯腈的可行性。2007年, 日本旭化成株式会社将一套年产7万吨的生产线改装成使用丙烷为原料生产丙烯腈。
丙烷氨氧化催化剂的活性组分主要采用V、Sb、P、Ga、Mo和Bi等, 除惰性元素以外的各族元素几乎均被用于催化剂改性。丙烷氨氧化反应完全由动力学因素控制, 不受热力学平衡限制。目前对丙烷直接氨氧化反应机理的认识尚不统一, 主要有两种, 一种认为丙烷先氧化脱氢生成丙烯, 再通过氨氧化反应生成丙烯腈; 另一种则认为丙烷先发生环化反应生成环化产物过渡态, 再与氨反应生成丙烯腈。
很多文献对丙烷氨氧化的催化剂以及催化反应进行了讨论, 提到的催化体系主要分为两种, 分别是基于具有金红石结构的V-锑酸盐和多组分钼酸盐(Mo/V/Nb/Te/O), 其中, Mo/V/Nb/Te/O体系最具潜力, 丙烯腈收率高达62%的[164, 165]。
7.5.3 甲苯及取代甲苯氨氧化
用于这类反应的催化剂有很多, 包括TiO2或
ZrO2负载的钒、多组分钼酸盐、掺杂的锑酸钒、负载的杂多酸等。很多文献对甲苯氨氧化过程中的催化剂表征、性能以及反应机理进行了讨论。以V2O5催化剂为例, 红外光谱表征表明, 反应为催化剂表面的铵离子与苯甲酸根离子的反应[166], 其中吸附苯甲胺和苯甲醛物种是反应的中间体[167]。V=NH或V— NHOH物种可能是催化剂表面进行插入反应的氮物种[168]。取代基对甲苯的氨氧化反应有明显影响, 吸电子取代基有利于提高腈的收率, 而给电子取代基会促进完全氧化, 对提高选择性不利。
7.5.4 小 结
混合金属氧化物催化的气相氨氧化是一类重要的工业生产过程, 其中最重要的丙烯氨氧化生产丙烯腈过程在最近40年取得了显著发展。当前生产丙烯腈的挑战在于用丙烷代替丙烯作原料。用于直接从丙烷到丙烯腈过程的技术和经济条件(丙烷和丙烯之间的价差)已经具备, 预计在不久的将来, 丙烯腈将越来越多地从丙烷制得。甲苯或取代甲苯氨氧化是成熟的反应, 已经被用于生产特殊用途的精细化学品。尽管已经实现了工业化, 但规模不大。
7.6 羰基化
在催化剂存在下, CO可与烯烃、卤代烷烃、饱和醇、醚、酯、胺、硝基化合物反应, 生成含羰基的化合物。烯烃与CO和氢气发生氢甲酰化反应生成醛, 加氢可得到醇[169]。全球采用此法生产醛、醇的产能年超过700万吨, 我国年产能达100万吨。
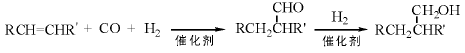
烯烃与CO和含有活泼氢的亲核试剂反应, 生成羧酸及其衍生物的过程为氢羧基化反应。
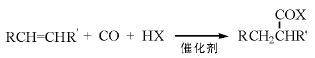
Monsanto公司以铑为主催化剂碘化物为助催化剂, 甲醇羰基化制乙酸工艺的反应, 是目前乙酸主要的生产方法。
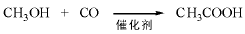
7.6.1 烯烃氢甲酰化
氢甲酰化反应是在过渡金属羰基化合物催化剂作用下, 烯烃与CO、H2生成比原烯烃多一分子的醛或醇的反应过程。氢甲酰化反应最早是由劳伦(Roelen O)于1938年在德国鲁尔化学公司从事费托合成中发现。
第一代氢甲酰化催化工艺均是以羰基钴为催化剂。Co2(CO)8首先转化成活性物种HCo(CO)4。HCo(CO)4极易分解为Co和CO, 需要维持高的压力(2030) MPa来保持活性物种的稳定性。
采用膦配体部分替代CO, 可提高正构醛的比例。这类配体稳定性较高, 可以在较低的压力下使用(10 MPa), 降低了设备的耐压要求。膦配体可使催化剂的稳定性增加, 但反应速率下降。另外, 该催化剂能够使醛进一步加氢生成醇, 产物中醛的量减少, 并使原料中的少量烯烃被加氢还原成烷烃, 使产品收率降低。
铑是比钴更具氢甲酰化催化活性的金属, 铑催化剂可以有效地在更温和的温度和压力下操作, 且醛收率高, 烯烃加氢和其他副反应都极大降低。使用未修饰的羰基铑催化剂, 正构醛含量低。采用三苯膦改性的铑催化剂, 反应条件温和、正构醛选择性高、醇含量低、反应速率快。20世纪70年代中期, 以HRh(CO)(PPh3)3为催化剂的氢甲酰化反应由美国联合碳化物公司(Union Carbide Corporation)实现工业化应用。然而, 铑的价格比钴高几千倍, 其回收、循环使用是该工艺的难点。
氢甲酰化反应伴随着很多副反应, 为了加快主反应和提高目标产品收率, 需要合理地控制反应条件。副反应主要有烯烃加氢、异构化、生成的醛被加氢生成醇。上述两种加氢反应, 与催化剂自身性质和反应条件有关。例如, 改性的钴催化剂加氢能力增强, 容易发生加氢副反应。氢气物质的量分数增加也会导致加氢副产物的增加。此外, 反应体系中烯烃量的减少还会引起甲烷化反应, 使反应体系温度升高。
大吨位的产品一般采用连续化工艺, 小吨位产品则采用间歇操作。生产过程包括催化剂制备、氢甲酰化反应、反应物料与催化剂分离、催化剂循环与再生、粗产品分离精制等。
催化剂制备一般在氢甲酰化反应器中进行, 将金属钴或钴化合物、烯烃一起送入氢甲酰化反应器, 一定反应条件下, 钴与CO和H2反应生成羰基钴。
氢甲酰化反应器通常为返混型, 间歇式反应可以采用釜式或塔式反应器。连续反应通常采用塔式反应器, 一般单塔即可。氢甲酰化反应是强放热过程, 需保证足够的冷却面积。
对于催化剂分离, 一般采用蒸馏分离方法。反应物料脱除气体后, 送入催化剂蒸馏分离塔, 将易挥发的产品蒸出, 含催化剂液体由塔底排出后经处理进入循环。目前, 鲁尔化学公司、壳牌公司、巴斯夫公司均采用此法。
7.6.2 烯烃氢羧基化
氢甲酰化反应中的氢分子, 可以采用水、醇、酸、胺等含活泼氢的亲核试剂替代, 生成羧酸及其衍生物, 这类反应称为氢羧基化反应。如果烯烃分子中含有活泼氢基团, 则生成相应的环状化合物。常采用镍、钴、钯、铹等金属的羰基化合物作为催化剂, 使用这种催化剂的氢羧基化反应称为Reppe羰基化。该催化剂的作用原理与催化氢甲酰化反应类似。这类催化剂还可催化炔烃的氢羧基化反应, 例如乙炔的氢羧基化合成丙烯酸、丙烯酸酯。
与甲酰化不同, 该反应还可采用硫酸、磷酸、HF、BF3等质子酸或Lewis酸作催化剂, 该体系的反应称为Koch反应。酸催化的历程是亲电加成过程, 反应第一步是质子进攻得碳正离子, 进攻的位置符合马克尼柯夫规则, 得到支链羧酸。此外, 碳正离子在反应过程中也可能发生异构化, 生成叔碳羧酸。金属羰基催化历程与氢甲酰化类似, 能够得到直链羧酸。直链羧酸与支链羧酸的比例与催化剂种类、反应条件等有关。
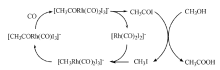
7.6.3 甲醇羰基化
饱和醇在水存在下与CO反应生成羧酸。伯醇和仲醇都可以发生类似反应, 例如, 以2-丁醇为原料可生成2-甲基丁酸, 无水条件下则生成酯。工业上甲醇合成乙酸就是采用羰基化路线。甲醇羰基化法占目前世界乙酸生产量的60%。工业上主要有Monsanto工艺和CativaTM工艺, Monsanto工艺以铑为主催化剂、碘化物为助催化剂。甲醇羰基化制乙酸工艺的反应条件为温度(150200) ℃、压力(36) MPa。乙酸收率基于甲醇为99%, 基于CO为85%。
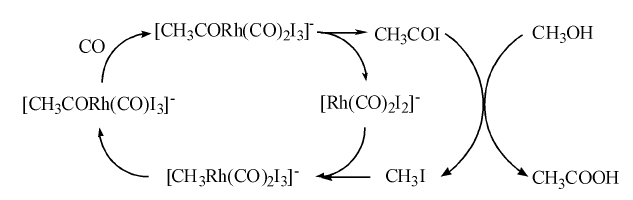 | 图 7-21 铑催化甲醇羰基化制乙酸的反应历程Figure 1 XRD patterns of the three-way catalysts |
铑催化甲醇羰基化制乙酸的反应历程如图7-21所示。其中CH3I与[Rh(CO)2I2]-的氧化加成反应为速控步骤。该方法的不足之处有:(1) 主催化剂铑的价格昂贵; (2) 反应体系中存在大量的水, 造成产物分离困难和投资增加; (3) 碘化物的存在会造成严重的设备腐蚀; (4) 催化剂稳定性不高和CO不足时, Rh易被氧化。
英国石油公司于1995年开发了以铱为主催化剂催化甲醇羰基化制乙酸的新工艺(CativaTM), 该催化体系具有较高的生产效率, 催化剂稳定性较高, 反应体系水浓度较低。催化剂的作用机理与羰基铑类似, 但速控步骤不同[170]。
7.6.4 卤代烷与CO的羰基化反应
卤素原子的电负性较强, 且与过渡金属具有较强的配位能力, 能进行配位体取代络合发生羰基化反应。亲核试剂可以是水、醇、胺等含活泼氢的分子。
丙二酸二乙酯是重要的有机合成中间体, 在染料、香料、磺酰脲类除草剂等生产中用途广泛, 目前, 主要以催化羰基化法生产。卤代乙酸乙酯和一氧化碳在乙醇溶剂中一步合成丙二酸二乙酯, 反应工艺简单、流程短、产物收率高、条件温和[171]。卤素比较活泼, 当采用卤代烯烃羰基化时, 羰基优先取代卤素, 双键保留。例如, 溴代丙烯在甲醇存在下, 发生羰基化生成丁烯酸甲酯。羰基化Suzuki偶联是合成酮类化合物的重要反应。采用苯硼酸为芳基化试剂, 碘代芳烃可与其发生羰基化Suzuki偶联反应。该反应的催化剂主要为Pd的配合物。末端炔也可作为烷基化试剂, 发生羰基化Sonogashira偶联反应, 生成α , β -不饱和炔酮化合物。
7.6.5 胺与CO的羰基化反应
在催化剂存在下, 胺可与CO作用生成N-甲酰基衍生物和脲素衍生物, 常用催化剂有锰、铜、钴的羰基化合物。在氧气存在下, 主要生成脲、氨基甲酸酯。
多种催化剂包括贵金属、非贵金属、非金属催化剂均可催化该类反应。如用于合成对称及不对称脲类化合物的Rh/C、[Ru(CO)3I3]NBu4、Co-Schiff碱、MnBr2、W(CO)6、纳米Au、PdI2/K、S和Se等催化剂体系; PdCl2/CuCl2、Pd/C-NaI等可用于氨基甲酸酯的合成。如果氨基的邻位有羟基, 则生成2-噁唑烷酮化合物。PdCl2(MeCN)2/CuI、PdI2/KI等均可催化该反应。
硝基化合物也可以作为该类反应的底物, 硝基被还原为氨基后, 可参与反应。例如采用Se催化剂、硝基苯为原料, 合成氨基甲酸酯。仲胺与叔胺也可与CO反应生成羰基化合物。
7.7 酯 化
7.7.1 酯化反应的类型
7.7.1.1 羧酸与醇的反应
一元醇与一元羧酸发生的反应最为简单, 只得到一种酯。二元羧酸与一元醇反应则生成单酯与双酯, 二者产率取决于原料配比。
多元羧酸与多元醇酯化, 可生成高分子聚酯。这类反应广泛应用于塑料及合成纤维的制备, 此外在涂料、黏合剂生产中也有应用。
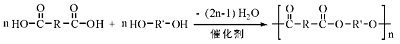
7.7.1.2 羧酸衍生物与醇反应
酰氯与醇生成酯的活性远高于羧酸, 反应为不可逆, 一般不需要催化剂即可进行。例如, 碳酸酯与磷酸酯的合成, 如直接采用碳酸或磷酸作为原料, 得不到理想的产物, 一般采用光气(COCl2)、POCl3与醇反应。采用酰氯作为原料成本高于羧酸, 只在特殊情况下使用, 如醇在高温下不稳定, 采用酰氯作为反应原料, 可在较低温度下反应。
酸酐也可替代酰氯, 第一步, 1 mol酸酐与1 mol醇生成1 mol酯与1 mol羧酸, 该步骤为不可逆过程; 第二步, 生成的羧酸与醇反应生成酯, 这一步骤为可逆过程, 且反应条件更为苛刻。
通常, 酯化反应的反应活性如下:
RCOCl> (RCO)2O> RCOOH
7.7.2 酯化反应历程与热力学
酸催化酯化是最常用的方法, 羧酸与伯醇、仲醇反应, 一般是双分子反应历程。酸催化酸与伯醇、仲醇的酯化历程如图7-22所示。
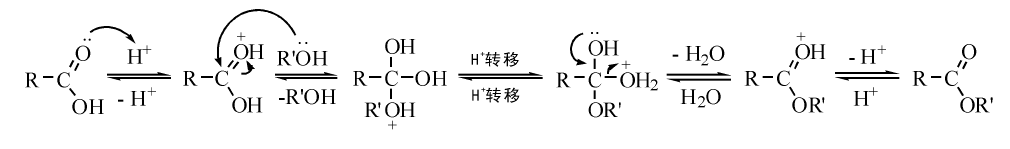 | 图 7-22 酸催化酸与伯醇、仲醇的酯化历程Figure 1 XRD patterns of the three-way catalysts |
叔醇的酯化历程与伯醇和仲醇的酯化历程不同, 多为单分子历程。此类反应为一级反应, 断开的是醇的烃氧键。例如叔丁醇的酯化, 即为该历程(见图7-23)。
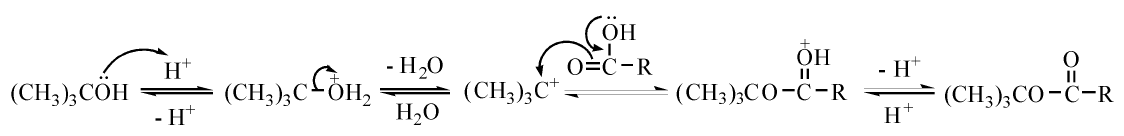 | 图 7-23 酸催化叔醇与酸的酯化历程Figure 1 XRD patterns of the three-way catalysts |
醇与羧酸的酯化反应热效应很小。水解与醇解过程类似, 热效应也不显著。这些过程的平衡常数与温度无关, 与醇和酸的结构均有关, 但醇的影响更为显著。液相酯化时平衡常数低(通常< 5), 通常可将反应产物水或酯蒸馏出以促进反应进行。也可采用某一原料过量的方法, 促使另一种反应物更多转化。
7.7.3 酯化反应的催化剂
酯化反应采用的催化剂主要有无机酸、有机酸、杂多酸及固载化的杂多酸、强酸性离子交换树脂、固体超强酸及分子筛等, 硫酸、盐酸、磷酸等无机酸最为常用。硫酸酸性最强, 能溶于反应体系中, 反应条件较温和, 催化活性最高, 是传统的酯化反应催化剂。但硫酸具有氧化性, 易使产品色泽变深; 此外硫酸腐蚀性强, 排出的酸性废水易造成环境污染。盐酸容易和醇发生反应生成氯代烷副产物。磷酸酸性相对较弱, 反应速率慢。苯磺酸、对甲苯磺酸、甲磺酸等有机酸, 酸性比硫酸弱, 活性较硫酸低, 但没有氧化性, 其中对甲苯磺酸最为常用, 如用于乙酸异戊酯、硬脂酸乙二醇酯等制备。
杂多酸是由P、Si、Fe、Co等杂原子和Mo、W、V、Nb、Ta等原子按一定结构, 通过氧原子配位桥联组成的一类含氧多酸, 不但具有酸性, 而且有氧化还原能力, 其中以H3PW12O40· 28H2O磷钨酸最为常用。杂多酸活性较高, 用量为反应物的1%2%, 反应温度比硫酸略低。如USY负载的磷钨酸铯盐催化剂在苹果酯的合成反应中, 不仅表现出较高的催化活性, 而且显示出较好的重复使用性能。
强酸性离子交换树脂催化剂易与产品分离, 可循环使用, 不腐蚀设备, 已广泛应用于酯化反应过程, 最常用的是酚磺酸树脂和磺化聚苯乙烯树脂。离子交换树脂目前已工业化, 酸性、孔结构参数可根据需要选择不同型号的产品。如采用苯乙烯-二乙烯基苯共聚磺化物作为催化剂, 可合成乙酸丁酯, 达到采用硫酸催化剂的水平, 催化剂寿命在500 h以上[172]。
固体超强酸因具有强酸性以及固体催化剂的特点, 倍受重视, 其中S
沸石分子筛具有很宽的可调变的酸中心和酸强度, 能够满足不同的酸催化反应的活性要求, 此外, 其比表面积大、孔分布均匀、孔径可调变、对反应原料和产物有良好的择形选择性。但分子筛具有脱水作用, 能够使醇脱水生成烯烃和醚, 因而反应时温度需严格控制。
目前报道的能够催化酯化反应的分子筛主要有H-ZSM-5、HY、Hβ 、H-MOR、磷酸铝分子筛等。以H-ZSM-5为催化剂, 合成乙酸正丙酯, 醇酸比为1, 110 ℃反应1 h, 产品收率约85%[174]。在间歇反应器中, 以磷酸铝分子筛催化酯化合成丙酸丁酯, (124133) ℃反应5 h, 酯收率可达85%, 催化剂重复使用4次, 活性降至79%[175]。
7.7.4 酯化工艺
酯化反应是一平衡反应, 从反应体系中分离出水, 促使反应向生成酯的方向进行。反应器可分为连续式和间歇式。当生产规模不大时, 常采用间歇式反应器, 反应器各部分组成与温度、物料停留时间均一致, 随着反应进行, 反应速率逐渐减慢, 可以逐渐提高反应温度。通常采用的间歇式反应器为带有搅拌和热交换的釜式反应器。连续操作的反应器类型较多, 主要有管式反应器、串联釜式反应器、塔式反应器等。可根据反应的动力学特征、传热、传质要求等选择合适的反应器。
7.8 展 望
“ 化石资源” 在石油化工和精细化工行业中的统治地位牢不可破, 但自20世纪70年代爆发能源危机, 人们已经意识到, 以化石资源为来源的化学品生产面临着严重危机, 有限的化石资源终有一天会消耗殆尽。随着石油化工和精细化工工艺迅速发展, 不可避免带来环境污染问题。人们已经意识到化学品大规模生产付出了牺牲环境的沉重代价, 需要研发环保催化剂和环保新技术、新工艺, 才能给后人留下足够生存空间, 才能维持社会的稳定运行。今后发展趋势如下:
(1) 石油化工趋向于发展大型化装置, 整体向着新技术开发、节能、工艺优化和产品延伸等方向发展。应以环境友好为出发点, 注重环境保护和减少环境污染, 审视与改进传统的石油化工、精细化工生产过程, 淘汰污染严重的传统工艺, 规范工艺的环保指标要求, 尽量降低“ 三废” 排放, 创造出新的环境友好的生产过程。
(2) 设计开发新型催化剂, 提高反应的“ 原子经济性” 。催化剂的革新是石油化工技术进步与发展的推动力; 随着先进仪器技术的发展, 催化基础研究也快速发展, 对催化活性中心/活性相概念的认知上升到分子层面[176], 能够对很多以前的模糊推测进行新的解释。这也为更高效催化剂的开发带来新的启发。
(3) 以生物质资源作为可持续提供化工原料的来源。相对于化石资源储量有限且不可再生, 生物质资源储量丰富、再生时间短, 具有替代化石资源的巨大潜力。我国是农业大国, 具有生物质资源开发利用的优势, 每年农作物秸秆达7亿吨, 为生物质路线生产能源和能源化学品提供了原料来源。利用催化和生物技术, 以生物质及生物基平台化合物为原料, 制备原依靠“ 石化路线” 生产的能源化学品, 是先进催化技术与生物质资源优化交叉利用的重要突破点。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
| [169] |
|
| [170] |
|
| [171] |
|
| [172] |
|
| [173] |
|
| [174] |
|
| [175] |
|
| [176] |
|