







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
EISA表面修饰法制备抗烧结催化剂
引用本文
潘喜强, 杨向光, 曾清湖, 李玉洁. EISA表面修饰法制备抗烧结催化剂[J]. 工业催化, 2016,24(5): 60-66.
Pan Xiqiang, Yang Xiangguang, Zeng Qinghu, Li Yujie. Preparation of sintering-resistant catalysts via EISA surface modification method[J]. Industrial Catalysis, 2016,24(5): 60-66.
DOI:10.3969/j.issn.1008-1143.2016.05.012
Pan Xiqiang, Yang Xiangguang, Zeng Qinghu, Li Yujie. Preparation of sintering-resistant catalysts via EISA surface modification method[J]. Industrial Catalysis, 2016,24(5): 60-66.
Permissions
Copyright©2016, 《工业催化》编辑部
《工业催化》编辑部 所有
EISA表面修饰法制备抗烧结催化剂
作者简介:潘喜强,1984年生,男,湖北省郧县人,博士,从事能源与环境催化研究。
摘要
通过催化剂表面修饰控制催化剂表面活性组分颗粒的表面迁移能力,可以提高催化剂的抗烧结性能。通过蒸发诱导自组装 (EISA)法修饰催化剂表面制备抗烧结催化剂,并对其性能进行研究。将经过验证的可形成二维有序介孔薄膜的EISA起始溶液沉积在Pd@SiO2模型催化剂表面,采用TEM表征mSiO2@Pd@SiO2催化剂结构,证明了EISA表面修饰法的可行性。将EISA表面修饰法用于修饰Pd/Al2O3催化剂,通过表面涂覆的次数控制修饰层的厚度。XRD和甲烷燃烧活性结果表明,经过修饰的mSiO2@Pd/Al2O3催化剂耐高温性能提高,其中,涂覆两次后的催化剂表面PdO纳米颗粒晶粒尺寸增大程度最小,甲烷燃烧活性最好。EISA表面修饰法制备抗烧结催化剂是一种提高催化剂高温稳定性的有效方法。
关键词:
催化剂工程; 抗烧结催化剂; Pd/Al2O3; EISA表面修饰法; 甲烷燃烧
中图分类号:TQ426.6
文献标志码:A
文章编号:1008-1143(2016)05-0060-07
Preparation of sintering-resistant catalysts via EISA surface modification method
Abstract
The surface migration of active component particles on the catalyst surface can be impeded via surface modification,thus enhancing sintering-resistant performance of the catalysts.The sintering-resistant catalysts were prepared by surface modification via evaporation-induced self-assembly (EISA) route,and their sintering-resistant properties were studied.EISA initial solution that was proved to form 2D ordered mesoporous thin film was deposited onto the surface of Pd@SiO2 model catalyst.The structure characterization of mSiO2@Pd@SiO2 catalyst derived from TEM experiment proved the feasibility of EISA surface modification method.Then EISA surface modification method was applied to coat the surface of Pd/Al2O3 catalyst,and the thickness of modification layer was controlled by the coating times.The results of XRD and methane combustion activities indicated that the stability of mSiO2@Pd/Al2O3 catalysts via EISA surface modification was enhanced at high temperatures,for PdO nanoparticles kept almost constant in size and mSiO2@Pd/Al2O3 catalyst had highest methane combustion activities after surface coating two times.Therefore,EISA surface modification method is a very effective approach to prepare sintering-resistant catalysts with enhanced stability.
Keyword:
catalyst engineering; sintering-resistant catalyst; Pd/Al2O3; EISA surface modification method; methane combustion
催化剂热失活是一个物理化学过程, 包括烧结、化学转换以及蒸发过程等, 其中, 烧结是高温催化剂失活的一个共性问题。烧结使催化剂活性相或载体表面积降低, 主要是晶体生长的过程, 因此, 烧结会降低催化剂活性, 当反应为结构敏感性时还会降低选择性。烧结为热力学驱动过程, 甄开吉等[1]将失活机理分为两种:Ostwald熟化机理, 原子从一个颗粒表面转移至另一个颗粒表面, 转移的通道可以是通过蒸发或者固体表面; Brown运动机理, 颗粒通过迁移聚集烧结。控制表面迁移性能和降低颗粒的饱和蒸气压可以提高高温催化剂的稳定性。高温催化剂的活性组分主要选择高熔点金属, 因其相应的Tamman温度和Hü ttig温度较高, 饱和蒸气压低, 较稳定。控制催化剂表面活性组分颗粒的表面迁移能力是进一步提高催化剂高温稳定性的有效途径。
通过催化剂表面修饰控制催化剂表面活性组分颗粒的表面迁移能力, 可以孤立活性相粒子, 防止其在高温下迁移。Joo S H等[2]合成了一种核为纳米Pt壳为介孔SiO2的Pt@mSiO2核壳结构模型催化剂, 未包覆前, 纳米Pt在300 ℃以上保护剂分解后即会聚集烧结, 通过介孔SiO2无机材料壁保护后, 纳米Pt可以耐750 ℃高温。这种修饰方法有很多限制, 首先壳层材料必须具有热稳定性好、机械强度高、对活性相无毒害作用和有足够的孔道不影响传质的性能。符合这些条件的壳层材料较少, 且合成过程复杂, 试剂昂贵, 不适合大量生产。其次, 为了提高催化剂效率, 需要将活性相颗粒(核)制备的尽可能小, 大大增加了合成核壳结构催化剂的难度。其他类似结构的Ni@Zr
蒸发诱导自组装(EISA)法是一种制备有序介孔薄膜材料的简单方法[12, 13], 起始溶液中包含硅溶胶、表面活性剂、水和乙醇, 其中, 表面活性剂浓度远低于临界胶束浓度(c0≪cmc), 通过乙醇的不断蒸发, 逐渐增加表面活性剂浓度, 并驱使硅溶胶和表面活性剂胶束自组装形成有序介孔液晶相。
EISA法合成有序介孔薄膜可以精确控制薄膜厚度, 通过控制初始浓度可以制备从一个单层胶束到多层厚度[14, 15]。EISA的基底可以是三维, Xue Chunfeng等[16]通过改变基底合成一系列不同的二氧化硅-碳的介孔复合物, 将这种方法称为EISA涂覆法。Li Lele等[17]通过同样的方法合成了氧化铝-聚亚胺酯复合物。You Bo等[18]合成了介孔碳-SiO2复合物, 沉积在苯乙烯纳米球表面, 通过后处理得到纳米空心介孔碳球。EISA是基于溶胶-凝胶过程, 因为大部分的氧化物均可用溶胶-凝胶法合成[19], EISA法表面修饰可大大拓展表面修饰物种的种类, 适合应用于催化剂表面修饰。
Pd/Al2O3催化剂在高温催化反应中应用广泛, 如汽车尾气三效催化剂、甲烷燃烧催化剂和甲烷部分氧化反应等。本文利用EISA法制备模型催化剂mSiO2@Pd@SiO2, 验证EISA表面修饰法的有效性, 将这种方法用于修饰常用的Pd/Al2O3催化剂, 并考察表面修饰后Pd/Al2O3催化剂的抗烧结性能。
1 实验部分
1.1 催化剂制备
1.1.1 EISA表面修饰法制备模型催化剂
SiO2纳米球制备:SiO2纳米球通过Stö ber法合成[20]。配制9 mL氨水、12.83 g乙醇和24.75 g水的混合溶液A。配制4.5 mL正硅酸乙酯和35.93 g乙醇的混合溶液B。将溶液B加至溶液A中, 室温反应3 h, 得到SiO2球, 离心分离, 水洗两遍, 乙醇洗一遍, 配制成一定浓度的SiO2溶液。
APTES-SiO2纳米Pd的制备: 纳米Pd根据乙二醇还原法合成[21, 22]。溶液C:1.11 g PVP加入到120 mL乙二醇溶液中, 80 ℃搅拌溶解3 h。冷却至0 ℃, 加入NaOH调节pH至9~11。溶液D:0.20 g醋酸钯加入到50 mL二氧六环中搅拌溶解。将溶液D加入到溶液C中, Ar气氛保护下, 100 ℃反应2 h即得到纳米Pd。纳米Pd的纯化:将一定体积的纳米Pd溶液加至丙酮溶液中搅拌沉降, 然后将得到的纳米Pd重新分散到水溶液中配制成一定浓度溶液。
Pd@SiO2制备:将SiO2球用3-氨丙基三乙氧基硅烷(APTES)修饰, 将APTES-SiO2与纳米Pd溶液以不同比例混合, 选取最佳的混合比例。
SiO2薄膜制备:根据文献[12, 23]报道的方法合成。将2.08 g正硅酸乙酯、(5~10) g乙醇、0.1 g的HCl(2 mol· L-1)以及(0.5~1) g的H2O混合搅拌溶解2 h。将一定浓度的溴化十六烷三甲基铵乙醇溶液加入上述溶液中, 保证溴化十六烷三甲基铵与Si物质的量比为0.1~0.12, 充分搅拌30 min混合。40 ℃陈化15 min, 然后将溶液倒入培养皿中, 室温蒸发诱导自组装24 h。将得到的样品在马弗炉中550 ℃焙烧5 h, 即得有序介孔SiO2。
mSiO2@Pd@SiO2制备: 将制备的Pd@SiO2加入到合成SiO2薄膜过程中陈化后的溶液中, 然后过滤, 洗涤, 室温保持24 h, 550 ℃焙烧5 h。
1.1.2 EISA法表面修饰Pd/Al2O3催化剂
Pd/Al2O3催化剂通过柠檬酸钯前驱体的等体积浸渍法合成 [10]。1 g柠檬酸加入到5 g的2%硝酸钯溶液中, 形成柠檬酸钯的配合物前驱体溶液, 然后与5 g的10 nm γ -Al2O3混合。将浸渍后的样品110 ℃ 干燥过夜, 马弗炉350 ℃焙烧 5 h。
mSiO2@Pd/Al2O3制备:用Pd/Al2O3催化剂等体积浸渍EISA过程中陈化的SiO2溶液, 然后干燥, 重复浸渍3次, 通过对比选择最佳涂覆次数。
1.2 催化剂表征
热失重分析采用Mettler-Toledo公司TGA/DSC 1STARe型热分析仪, 样品在100 ℃恒温条件下失水后, 计算固含量。
小角X射线散射测试在奥地利安东帕公司SAXSpace型X射线散射仪上完成, 将粉末样品装入样品池, 真空状态曝光2 h收集信号, 工作电压40 kV, 工作电流50 mA, 铜靶, X射线波长0.154 nm。
XRD测试在德国布鲁克公司D8 Advance型X射线衍射仪上进行, CuKα , λ =0.154 06 nm, 工作电压40 kV, 工作电流20 mA, 步长0.02° , 扫描范围10° ~80° , 扫描速率5° · min-1。
SEM样品形貌由荷兰飞利浦仪器公司XL-30型场发射扫描电子显微镜测试, 样品制备好后在15 kV下进行检测。
TEM表征在日本电子株式会社JEM-2010透射电子显微镜上进行, 操作电压100 kV。
HRTEM表征在FEI公司Tecnai G2S-Twin场发射透射电子显微镜上进行, 操作电压200 kV。
1.3 催化剂性能评价
甲烷催化燃烧反应活性评价采用常压固定床石英管反应器, 反应器内径 6 mm, 长度40 cm。在线质谱仪(Hiden Analytical QIC-20)通过质核比m/z=15检测甲烷浓度变化。体积分数1%甲烷-空气混合气为反应原料, 空速为75 000 mL· (g· h)-1, 催化剂活性通过程序升温反应评价。
2 结果与讨论
2.1 mSiO2@Pd@SiO2的结构
图1为采用Stö ber法合成的SiO2球的SEM照片。由图1可以看出, 得到的SiO2球分散性非常好, 直径约为250 nm。TGA测试结果发现, SiO2球溶液的固含量为2.22%。
 | 图 1 合成的SiO2球的SEM照片Figure 1 SEM images of synthesized SiO2 nanospheres |
图2为以PVP为保护剂合成的纳米Pd的TEM照片。
 | 图 2 纳米Pd的TEM照片Figure 2 TEM images of Pd nanoparticles |
由图2可以看出, 纳米Pd颗粒粒径均匀, 平均为(2~3) nm, 且纳米Pd分散性较好, 没有出现团聚现象。
为了使纳米Pd均匀分散在SiO2球表面, 需要对SiO2球表面功能化。3-氨丙基三乙氧基硅烷常用于SiO2球表面的修饰, 使其表面带氨丙基基团。本文中3-氨丙基三乙氧基硅烷用量按照在SiO2球表面增加2.5个原子层厚度的理论量修饰, 在Stö ber法合成的SiO2球溶液中, 加入20 μ L的3-氨丙基三乙氧基硅烷继续反应10 h, 具体计算过程见参考文献[24, 25]。考察了3-氨丙基三乙氧基硅烷修饰的SiO2和纳米Pd比例对Pd@SiO2表面分散性的影响。纳米Pd溶液和氨基化的SiO2溶液的体积比分别为0.01、0.03、0.7和0.14, 二者混合后搅拌并超声分散, 然后离心分离, 用去离子水洗3遍。不同比例纳米Pd和SiO2负载后Pd/SiO2催化剂的TEM照片见图3。
 | 图 3 不同体积比纳米Pd和SiO2负载后Pd/SiO2催化剂的TEM照片Figure 3 TEM images of Pd/SiO2 catalysts loaded with different volume ratios of nanoparticles Pd to SiO2 |
由图3可以看出, 负载体积比为0.01和0.03时, 在SiO2球表面几乎看不到纳米Pd颗粒, 表明负载量太小; 当负载体积比增至0.7时, 能够明显观察到分散的纳米Pd颗粒, 继续增大体积比至0.14, 未观察到明显的团聚现象。
纳米Pd和SiO2体积比为0.14时的Pd/SiO2催化剂TEM和HRTEM照片如图4所示。由图4可以看出, 纳米Pd颗粒负载后形貌得到很好的保持, 同时纳米Pd颗粒均匀分散在SiO2表面, 未发生团聚。表明3-氨丙基三乙氧基硅烷修饰的效果非常均匀, 氨基均匀分散在SiO2球表面, 同时纳米Pd颗粒没有过量, 无团聚现象。因此, 选用纳米Pd和SiO2体积比为0.14合成模型催化剂Pd@SiO2。
 | 图 4 纳米Pd和SiO2体积比为0.14时Pd/SiO2催化剂的TEM和HRTEM照片Figure 4 TEM and HRTEM images of Pd/SiO2 catalyst with nanoparticles Pd/SiO2 volume ratio of 0.14 |
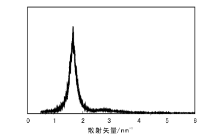
为了合成mSiO2@Pd@SiO2, 首先需要保证采用的EISA表面修饰法能够合成SiO2介孔薄膜, 然后采用诱导蒸发前的SiO2溶胶涂覆Pd@SiO2。EISA表面修饰法制备的SiO2介孔薄膜SAXS表征如图5所示。由图5可以看出, 合成的SiO2介孔薄膜介孔孔道长程有序, 经计算孔径为4.7 nm。
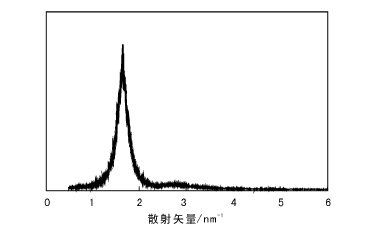 | 图 5 介孔SiO2薄膜的SAXS图Figure 5 SAXS profile of mesoporous SiO2 thin film |
用EISA起始溶液修饰Pd@SiO2催化剂, 修饰一次后, mSiO2@Pd@SiO2催化剂的HRTEM照片如图6所示。
 | 图 6 mSiO2@Pd@SiO2催化剂的HRTEM照片Figure 6 HRTEM image of mSiO2@Pd@SiO2 catalyst |
由图6可以看出, 起始溶液修饰的厚度约为2 nm, 孔道垂直于SiO2表面, 与文献[18]一致。证明EISA表面修饰法修饰催化剂表面可行, 将这种方法应用于Pd/Al2O3 表面修饰, 制备mSiO2@Pd/Al2O3催化剂, 并考察修饰后催化剂的稳定性。
2.2 mSiO2@Pd/Al2O3催化剂的抗烧结性能
图7为催化剂经过不同温度焙烧后的甲烷燃烧活性。由图7a可以看出, Pd/Al2O3-350催化剂的甲烷燃烧T10、T50和T90温度分别为309 ℃、335 ℃和382 ℃, 活性非常好, 能够在400 ℃以内完全转换甲烷。随着焙烧温度升高, Pd/Al2O3-750的T10、T50和T90温度分别为322 ℃、360 ℃和454 ℃, Pd/Al2O3-850的T10、T50和T90温度分别为377 ℃、424 ℃和580 ℃, 所有温度均升高, 表明催化剂的催化活性明显降低。尽管Pd/Al2O3初活性很高, 但抗烧结能力差, 活性难以保持。
用EISA表面修饰法修饰Pd/Al2O3催化剂, 根据修饰次数的不同分别标记为EISA0-Pd/Al2O3、EISA1-Pd/Al2O3、EISA2-Pd/Al2O3和EISA3-Pd/Al2O3。将样品分别在550 ℃、750 ℃和850 ℃焙烧5 h, 观察催化剂活性的变化。由图7b可以看出, 550 ℃焙烧后, 活性顺序为:EISA0-Pd/Al2O3> EISA1-Pd/Al2O3> EISA2-Pd/Al2O3> EISA3-Pd/Al2O3。表明表面修饰降低了催化剂活性, EISA修饰1~2次对活性影响较小, 修饰3次后, 活性严重下降, 可能是影响催化剂传质过程所致。
由图7c可见, 750 ℃焙烧后活性顺序不变, 但EISA1-Pd/Al2O3、EISA2-Pd/Al2O3和EISA0-Pd/Al2O3的T10和T50温度基本相同, T90有差别, 考虑到EISA1-Pd/Al2O3和EISA2-Pd/Al2O3的活性组分含量略低, 表明修饰对于提高抗烧结性能有效。
由图7d可以看出, 焙烧温度提高至850 ℃, 活性顺序为:EISA2-Pd/Al2O3> EISA0-Pd/Al2O3> EISA3-Pd/Al2O3> EISA1-Pd/Al2O3, 表明EISA法修饰两次可以延迟高温烧结。
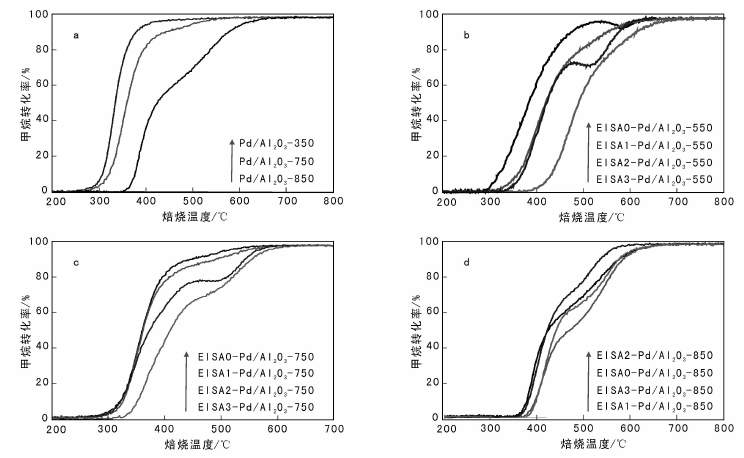 | 图 7 催化剂经过不同温度焙烧后的甲烷燃烧活性Figure 7 Methane combustion performance of the catalysts at different calcination temperatures |
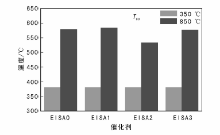
不同催化剂850 ℃焙烧5 h后与Pd/Al2O3-350的T90温度对比如图8所示。由图8可以看出, 850 ℃焙烧后, EISA0-Pd/Al2O3、 EISA1-Pd/Al2O3、EISA2-Pd/Al2O3和EISA3-Pd/Al2O3的甲烷燃烧T90温度分别为578 ℃、584 ℃、533 ℃和576 ℃。EISA2-Pd/Al2O3的抗烧结效果最好, T90温度最低, 与Pd/Al2O3-350的T90温度差距最小。因此用SiO2修饰时, EISA表面修饰法修饰两次效果较好。EISA表面修饰法修饰的效果理论上比实验效果更好, 因为修饰后催化剂质量增加, 活性相的绝对量降低, 修饰后催化剂比活性比实验值高。另外由于表面修饰物SiO2本身并不耐高温, 表面修饰层先烧结会导致催化剂失活, 用高温材料效果更好。
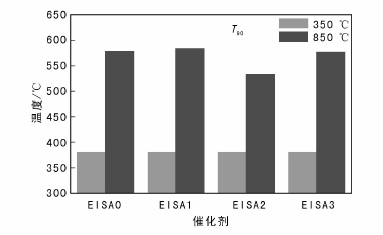 | 图 8 催化剂修饰前后T90温度Figure 8 T90 temperatures of the catalysts before and after surface modification |
2.3 催化剂烧结过程表征
采用TEM表征Pd/Al2O3催化剂在不同温度焙烧后的PdO纳米颗粒粒径, 结果如图9所示。由图9可以看出, 初始制备的催化剂Pd/Al2O3-350 ℃的PdO平均粒径约为3 nm, 且尺寸均匀, 表明柠檬酸法可以很好地控制钯催化剂的粒径, 与文献[10]结
果一致。550 ℃焙烧5 h后, PdO的粒径增至约7 nm。850 ℃焙烧5 h后, PdO的粒径增加至约11 nm, 可见Pd/Al2O3抗烧结性能较差, 与图7a中活性随着焙烧温度增加而降低的结果一致。
 | 图 9 Pd/Al2O3催化剂不同温度焙烧后TEM照片Figure 9 TEM images of Pd/Al2O3 catalyst after calcination at different temperatures |
修饰后的催化剂抗烧结性能通过XRD进行表征, 催化剂550 ℃和 850 ℃焙烧后XRD图见图10。
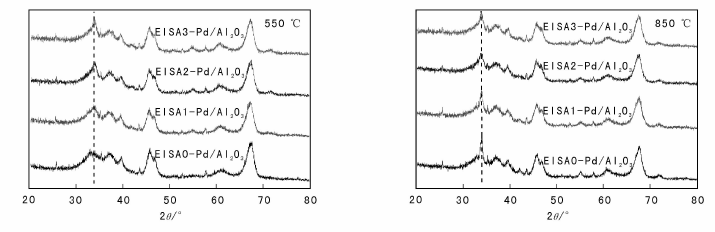 | 图 10 催化剂550 ℃和 850 ℃焙烧后XRD图Figure 10 XRD patterns of catalysts calcined at 550 ℃ and 850 ℃ |
由图10可见, 不同催化剂在550 ℃焙烧5 h后, 均在33.8° 处出现PdO衍射峰, 对应PdO的(101)晶面, 采用谢乐公式计算出PdO粒径, 所有催化剂中PdO粒径约为5.5 nm, EISA修饰后的催化剂中PdO粒径略有增加, 可能与溶液干燥过程中引起PdO粒径迁移有关。不同催化剂在850 ℃焙烧5 h后, EISA0-Pd/Al2O3、EISA1-Pd/Al2O3、EISA2-Pd/Al2O3和EISA3-Pd/Al2O3中PdO粒径分别为8.6 nm、8.5 nm、6.1 nm和7.3 nm, 表明随着焙烧温度升高, PdO颗粒尺寸逐渐增大。修饰后催化剂与未修饰催化剂相比, PdO粒径增大幅度明显降低, 且修饰次数越多, 粒径越小, 但修饰两次的最佳, 这可能与SiO2与Al2O3之间的固相反应有关; 修饰3次后, SiO2过量, SiO2本身的烧结较明显, 需要寻找更好的修饰材料。EISA2-Pd/Al2O3的PdO粒径小于EISA0-Pd/Al2O3, 这很好地解释了EISA2-Pd/Al2O3的甲烷燃烧活性比EISA0-Pd/Al2O3好的原因(图7d)。
3 结论
(1) 将能够形成有序介孔SiO2薄膜的前驱体溶液与Pd@SiO2催化剂混合, 通过EISA表面修饰法将有序介孔SiO2薄膜沉积在Pd@SiO2催化剂表面。TEM结果表明形成了mSiO2@Pd@SiO2结构, 证明EISA表面修饰法可以很好的将氧化物沉积在催化剂表面, 达到表面修饰效果。
(2) Pd/Al2O3催化剂具有较好的甲烷燃烧催化活性, 但不抗烧结, 随着焙烧温度升高, 催化活性下降明显。通过EISA表面修饰法修饰Pd/Al2O3后, 发现mSiO2@Pd/Al2O3催化剂的抗烧结性能提高, 850 ℃焙烧后的催化剂活性优于未修饰的催化剂。XRD和TEM分析发现, EISA修饰后的催化剂可以抑制PdO粒径的增大。其中, EISA修饰两次后的催化剂EISA2-Pd/Al2O3具有最佳的抗烧结表现。
(3) EISA表面修饰法修饰催化剂能够很好地控制修饰层的厚度和扩散性能, 并且将修饰层的种类扩展至多种氧化物。因此, 通过EISA表面修饰法制备抗烧结催化剂是提高催化剂稳定性的有效方法。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|