{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cu-Ce-O/γ-Al2O3的CO氧化性能
引用本文
储丽丽, 孔令朋, 张传驹, 陈亮, 靳广洲. Cu-Ce-O/γ-Al2O3的CO氧化性能 [J]. 工业催化, 2017,25(11): 28-33.
Chu Lili, Kong Lingpeng, Zhang Chuanju, Chen Liang, Jin Guangzhou. The performances of CO oxidationover Cu-Ce-O/γ-Al2O3 catalyst [J]. Industrial Catalysis, 2017,25(11): 28-33.
DOI:10.3969/j.issn.1008-1143.2017.11.006
Chu Lili, Kong Lingpeng, Zhang Chuanju, Chen Liang, Jin Guangzhou. The performances of CO oxidationover Cu-Ce-O/γ-Al2O3 catalyst [J]. Industrial Catalysis, 2017,25(11): 28-33.
Permissions
Copyright©2017, 《工业催化》编辑部
《工业催化》编辑部 所有
Cu-Ce-O/γ-Al2O3的CO氧化性能
作者简介:储丽丽,1991年生,女,黑龙江省鹤岗市人,在读硕士研究生。
摘要
采用柠檬酸络合法制备一系列不同铜铈比的Cu-Ce-O/γ-Al2O3催化剂,用XRD、H2-TPR对其进行表征,采用连续固定床微反装置对Cu-Ce-O/γ-Al2O3催化剂CO催化氧化活性进行评价。结果表明,Cu-Ce-O/γ-Al2O3催化剂的XRD图谱中除归属于γ-Al2O3的晶相峰外,还出现CuO和CeO2的晶相峰。高温水热引起活性组分CeO2的晶粒聚集、长大和尖晶石结构CuAl2O4物质的生成;CuO-CeO2之间的共生共存与相互作用,使得Cu-Ce-O/γ-Al2O3催化剂中具有非完整结构的[C
关键词:
催化化学; XRD; H2-TPR; 氧化铜; 氧化铈; CO催化氧化; Cu-Ce-O/γ-Al2O3催化剂
中图分类号:O643.32;TE624.9+4
文献标志码:A
文章编号:1008-1143(2017)11-0028-06
The performances of CO oxidationover Cu-Ce-O/γ-Al2O3 catalyst
Abstract
A series of Cu-Ce-O/γ-Al2O3 catalysts with different Cu/Ce ratio were prepared by citrate method and characterized by XRD,H2-TPR and testedactivityfor oxidation of carbon monoxide in continuous micro-fixed bed reactor.The results showed that there werecrystalline peaks of CeO2 and CuO in Cu-Ce-O/γ-Al2O3 catalysts besides γ-Al2O3.High temperature hydrothermal conditioninduced aggregation and grain growth of active components and the formation of CuAl2O4 in the Cu-Ce-O/γ-
Keyword:
catalytic chemistry; XRD; H2-TPR; copper oxide; ceria oxide; CO catalytic oxidation; Cu-Ce-O/γ-Al2O3 catalyst
原油重质化趋势加大, 催化裂化反应过程中焦炭产率增加, 使得催化裂化再生过程的烧焦负荷加大[1, 2]。催化裂化烧焦再生过程中添加CO助燃剂目的是有效脱除再生烟气中的CO, 避免再生烟气尾燃现象的发生[3, 4]。目前国内外广泛使用的CO助燃剂多是以贵金属Pt、Pd等为活性组分的贵金属助燃剂。贵金属铂助燃剂虽能有效促进催化裂化催化剂的烧焦再生, 减少烟气中CO含量, 但由于资源所限, 价格昂贵, 且使用铂助燃剂会大幅度增加再生器烟气中NOx的排放量, 环境污染加剧[5, 6]。在废气净化领域, Cu、Mn、Cr、Co、W等的氧化物或复合氧化物表现出较高的CO氧化活性[7, 8, 9, 10], 其中, CuO被认为是最有可能替代贵金属的催化活性材料。但与贵金属Pt、Pd催化剂相比, CuO的催化氧化活性相对偏低, 如何提高其催化氧化活性使其与贵金属相近, 一直受到国内外学者的关注[11, 12, 13]。CeO2因其独特的储/释氧能力使得CuO-CeO2复合氧化物催化剂表现出很好的CO氧化活性, 是贵金属催化材料的最佳替代品[14, 15, 16]。
本文采用改进的柠檬酸络合法制备二相共生共存的Cu-Ce-O/γ -Al2O3复合氧化物催化剂, 采用XRD、TPR对其物相结构和表面性质进行表征, 结合CO氧化活性的研究, 揭示二相共生共存效应对其CO催化氧化性能的影响。
1 实验部分
1.1 原料与试剂
Cu(NO3)2· 3H2O、Ce(NO3)3· 6H2O, 分析纯, 国药集团化学试剂有限公司; 柠檬酸, 分析纯, 北京化工厂; γ -Al2O3, 工业级, 山东铝业公司研究院; 5.0%H2/Ar标准气, 北京市北温气体制造厂; 6.0%CO+3.64%O2/He标准气, 北京市北温气体制造厂。
1.2 Cu-Ce-O/γ -Al2O3催化剂的制备
将相应的Ce3+溶液和Cu2+溶液按等体积浸渍到γ -Al2O3上, 经陈化、烘干、热解、活化制得实验用CuO/γ -Al2O3和CeO2/γ -Al2O3催化剂。按Cu与Ce物质的量比9∶ 1、8∶ 2、7∶ 3、6∶ 4、5∶ 5、4∶ 6、3∶ 7、2∶ 8、1∶ 9取Ce3+溶液和Cu2+溶液配制成混合溶液, 加入适量柠檬酸, 搅拌使其全部溶解后, 静置过夜。将上述混合溶液等体积浸渍到γ -Al2O3上, 经陈化、烘干、热解、活化制得Cu-Ce-O/γ -Al2O3样品, 标记为Cu-Ce-O/γ -Al2O3-mn, 其中mn为样品中Cu与Ce物质的量比。
1.3 样品的表征
采用日本理学公司D/max-2600-PC型X射线衍射仪对制备的催化剂样品进行物相分析, CuKα , 工作电压40 kV, 工作电流100 mA, 石墨单色器, 闪烁计数器, 入射狭缝1° , 接收狭缝0.3 mm, 扫描速率1° · min-1, 步幅0.02° , 扫描范围8° ~80° 。样品的TPR分析在泛泰仪器公司的FINESORB-3010程序升温化学吸附仪上进行, 在石英管中装入0.2 g(40~60)目的样品, 400 ℃预处理40 min, 然后降至室温, 用5.0%H2-Ar标准气吹扫30 min至检测器基线稳定, 由室温开始程序升温至700 ℃并稳定30 min, 升温速率为10 ℃· min-1, 热导池检测器。
1.4 CO催化氧化性能评价
CO催化氧化评价在连续固定床微反装置进行, 反应尾气用北京北分瑞利分析仪器有限公司SP-3420A气相色谱仪进行检测, TCD检测器, 5A分子筛柱。在反应温度下, 通入反应混合气, 1 h后开始测量, 取5次结果的平均值计算催化剂样品对CO氧化性能的转化率。
2 结果与讨论
2.1 XRD
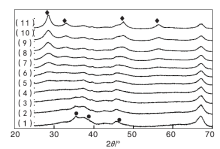
实验制备的CuO/γ -Al2O3、CeO2/γ -Al2O3和Cu-Ce-O/γ -Al2O3-mn系列催化剂的XRD表征结果见图1。
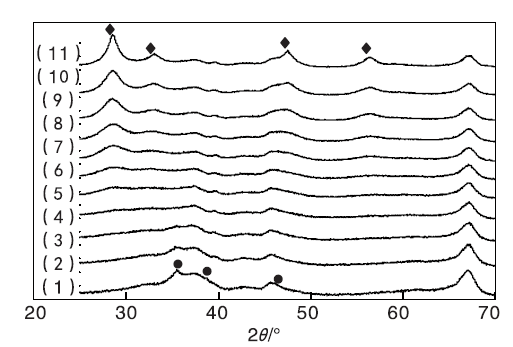 | 图1 Cu-Ce-O/γ -Al2O3-mn催化剂XRD图Figure 1 XRD patterns of the Cu-Ce-O/γ -Al2O3-mn samples (1)CuO/γ -Al2O3; (2)Cu-Ce-O/γ -Al2O3-91; (3)Cu-Ce-O/γ -Al2O3-82; (4)Cu-Ce-O/γ -Al2O3-73; (5)Cu-Ce-O/γ -Al2O3-64; (6)Cu-Ce-O/γ -Al2O3-55; (7)Cu-Ce-O/γ -Al2O3-46; (8)Cu-Ce-O/γ -Al2O3-37; (9)Cu-Ce-O/γ -Al2O3-28; (10)Cu-Ce-O/γ -Al2O3-19; (11)CeO2/γ -Al2O3 |
图1以看出, CuO/γ -Al2O3样品的XRmD图中, 除归属于γ -Al2O3的衍射峰外, 在35.544° 、38.709° 和46.260° 出现了归属于单斜晶系CuO的(11
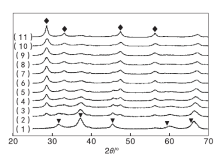
为考察实验制备的Cu-Ce-O/γ -Al2O3-mn催化剂的高温水热性能, 对制备的新鲜催化剂置于788 ℃、100%水蒸汽条件下进行高温水热老化处理12 h, 水热老化处理后催化剂的XRD图谱见图2。
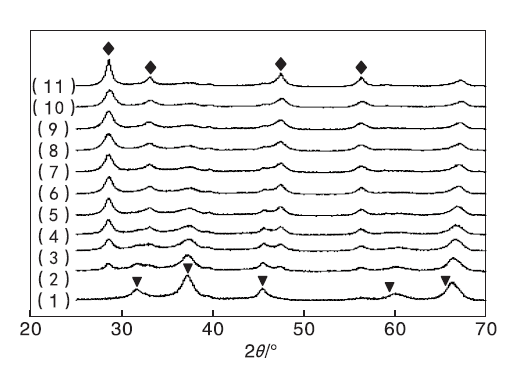 | 图 2 Cu-Ce-O/γ -Al2O3-mn催化剂水热后XRD图Figure 2 XRD patterns of the the hydrothermal samples of Cu-Ce-O/γ -Al2O3-mn (1)CuO/γ -Al2O3; (2)Cu-Ce-O/γ -Al2O3-91; (3)Cu-Ce-O/γ -Al2O3-82; (4)Cu-Ce-O/γ -Al2O3-73; (5)Cu-Ce-O/γ -Al2O3-64; (6)Cu-Ce-O/γ -Al2O3-55; (7)Cu-Ce-O/γ -Al2O3-46; (8)Cu-Ce-O/γ -Al2O3-37; (9)Cu-Ce-O/γ -Al2O3-28; (10)Cu-Ce-O/γ -Al2O3-19; (11)CeO2/γ -Al2O3 |
由图2可以看出, CuO/γ -Al2O3催化剂的XRD图中, 归属于单斜晶系CuO的特征衍射峰消失, 取而代之的是位于31.294° 、36.867° 、44.855° 、59.420° 和65.292° 处的衍射峰, 分别归属于立方晶系尖晶石结构CuAl2O4的(220)、(311)、(400)、(511)和(440)晶面。由于此处的衍射峰与归属于γ -Al2O3的31.936° 、37.603° 、45.788° 、60.457° 、66.761° 衍射峰部分重叠, 使得归属于立方晶系尖晶石结构CuAl2O4的衍射峰位置向高角度偏移。此部分衍射峰的出现, 表明CuO/γ -Al2O3催化剂在788 ℃、100%水蒸汽水热处理12 h的过程中发生活性组分CuO与载体γ -Al2O3的相互作用, 生成具有尖晶石结构的CuAl2O4物质。CeO2/γ -Al2O3催化剂的XRD图中, 归属于立方晶系CeO2的衍射峰依然存在。与图1相比, 图2中水热后CeO2/γ -Al2O3催化剂的XRD衍射峰强度增强, 峰形变窄, 表明高温水热引起活性组分CeO2的晶粒聚集、长大。图2中的衍射峰与CeO2的特征衍射峰位置相对应, 同样存在其衍射峰强度增强, 峰形变窄的现象, 进一步验证了水热引起活性组分CeO2的晶粒聚集、长大。水热后Cu-Ce-O/γ -Al2O3-mn催化剂的XRD图中归属于立方晶系尖晶石结构CuAl2O4的衍射峰强度减弱, 且随Cu与Ce物质的量比的减小明显减弱, 且峰形变宽, 表明CuO-CeO2的二相共生共存、相互作用有利于减少高温水热环境下活性组分的聚集和晶粒长大, 有利于减少尖晶石结构CuAl2O4物质的生成。
2.2 H2-TPR
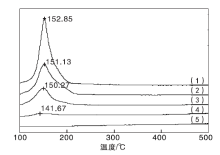
实验制备的CuO/γ -Al2O3、CeO2/γ -Al2O3和Cu-Ce-O/γ -Al2O3-mn系列催化剂的H2-TPR测试结果见图3。
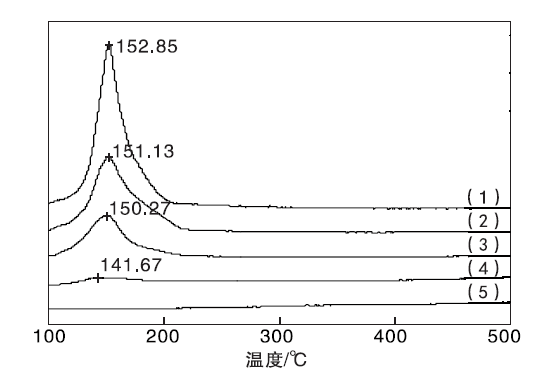 | 图3 Cu-Ce-O/γ -Al2O3-mn催化剂的H2-TPR图谱Figure 3 H2-TPR profiles of the Cu-Ce-O/γ -Al2O3-mn catalysts (1)CuO/γ -Al2O3; (2)Cu-Ce-O/γ -Al2O3-82; (3)Cu-Ce-O/γ -Al2O3-64; (4)Cu-Ce-O/γ -Al2O3-28; (5)CeO2/γ -Al2O3 |
图3中出现的均为CuO的H2-TPR还原峰, 没有观察到CeO2的还原峰, 表明CeO2在此温度范围不能被还原, Cu-Ce-O/γ -Al2O3-mn催化剂起氧化还原作用的主要是CuO相。CuO/γ -Al2O3催化剂的H2-TPR还原峰温度为152.85 ℃。与之相比, Cu-Ce-O/γ -Al2O3-82催化剂的还原峰温度降至151.13 ℃, Cu-Ce-O/γ -Al2O3-28催化剂的还原峰温度位于141.67 ℃。随着Cu与Ce物质的量比减小, Cu含量下降, Ce含量增多, Cu-Ce-O/γ -Al2O3-mn催化剂中归属于CuO的还原峰温度逐渐向低温区偏移, 表明CeO2的加入促进了CuO的还原。Cu-Ce-O/γ -Al2O3-mn催化剂制备过程中, CuO-CeO2之间的共生共存、强相互作用, Ce3+/Ce4+之间的可变价转换, 使得Cu-Ce-O/γ -Al2O3-mn催化剂的表相CuO中具有非完整结构的[C
2.3 CO催化氧化性能
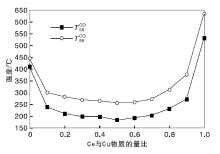
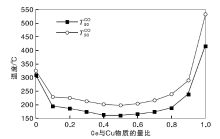
Cu-Ce-O/γ -Al2O3催化剂的CO催化氧化性能评价实验用CO转化率为50%和90%时的温度
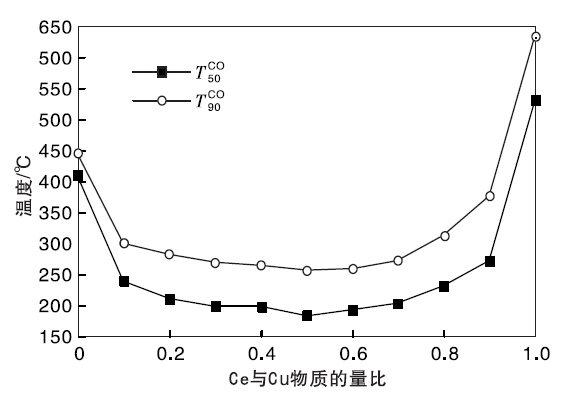 | 图4 新鲜Cu-Ce-O/γ -Al2O3-mn催化剂CO催化氧化性能Figure 4 The CO catalytic oxidation performance of Cu-Ce-O/γ -Al2O3-mn fresh catalysts |
由图4可以看出, 新鲜CuO/γ -Al2O3催化剂的
实验制备的Cu-Ce-O/γ -Al2O3-mn新鲜催化剂高温水热老化处理得到的水热样品的CO氧化性能评价结果见图5。
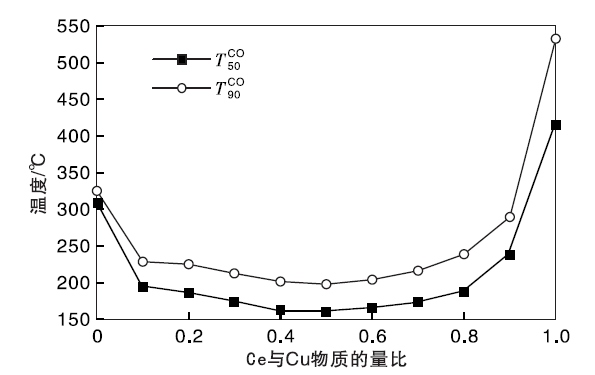 | 图5 水热老化Cu-Ce-O/γ -Al2O3-mn催化剂CO催化氧化性能Figure 5 The CO catalytic oxidation performance of Cu-Ce-O/γ -Al2O3-mn catalystsby hydrothermal treatment |
由图5可以看出, 高温水热老化处理后Cu-Ce-O/γ -Al2O3-mn催化剂的
3 结 论
(1) Cu-Ce-O/γ -Al2O3-mn催化剂的XRD图中除归属于γ -Al2O3的晶相峰外, 还出现了CuO和CeO2的晶相峰。随Cu与Ce物质的量比减小, Cu-Ce-O/γ -Al2O3-mn催化剂的XRD图中归属于CeO2的衍射峰逐渐增强, 而归属于CuO的特征衍射峰强度则逐渐减弱直至消失。至Cu与Ce物质的量比降至7∶ 3制备的Cu-Ce-O/γ -Al2O3-73样品中几乎观测不到归属于CuO的XRD特征衍射峰存在。高温水热引起活性组分CeO2的晶粒聚集、长大, 引起了活性组分CuO与载体γ -Al2O3的相互作用生成尖晶石结构的CuAl2O4物质; CuO-CeO2的二相共生共存、相互作用有利于减少高温水热环境下活性组分的聚集、晶粒长大及尖晶石结构CuAl2O4物质的生成。
(2) CuO-CeO2之间的共生共存、相互作用, 使得制备的Cu-Ce-O/γ -Al2O3-mn催化剂中表相CuO中具有非完整结构的[C
(3) CuO-CeO2二相共生共存、相互作用, 有利于非完整结构的[C
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|