
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MoO3-CeO-Co3O4催化剂CO催化氧化及耐硫性能
引用本文
邢聪聪, 李坚, 梁文俊, 樊星, 史蕊. MoO3-CeO-Co3O4催化剂CO催化氧化及耐硫性能 [J]. 工业催化, 2017,25(6): 39-42.
Xing Congcong, Li Jian, Liang Wenjun, Fan Xing, Shi Rui. Performance of MoO3-CeO-Co3O4 catalysts for CO catalytic oxidation and sulfur resistance [J]. Industrial Catalysis, 2017,25(6): 39-42.
Doi:10.3969/j.issn.1008-1143.2017.06.009Xing Congcong, Li Jian, Liang Wenjun, Fan Xing, Shi Rui. Performance of MoO3-CeO-Co3O4 catalysts for CO catalytic oxidation and sulfur resistance [J]. Industrial Catalysis, 2017,25(6): 39-42.
Permissions
Copyright©2017, 《工业催化》编辑部
《工业催化》编辑部 所有
MoO3-CeO-Co3O4催化剂CO催化氧化及耐硫性能
作者简介:邢聪聪,1990年生,女,山东省济南市人,在读硕士研究生。
摘要
采用共沉淀法和浸渍法联用制备不同Mo质量分数的 xMoO3-6CeO-Co3O4催化剂,测试催化剂催化氧化CO效率及其耐硫性能,并对催化剂进行BET、SEM、FT-IR和H2-TPR等表征。结果表明,MoO3的添加可以提高催化剂低温活性,5.61MoO3-6CeO-Co3O4催化剂低温活性最佳,40 ℃时CO去除率达98%,耐硫性能达90 min。
关键词:
催化化学; CO催化; 低温活性; 耐硫性能; MoO3
中图分类号:O643.36
文献标志码:A
文章编号:1008-1143(2017)06-0039-04
Performance of MoO3-CeO-Co3O4 catalysts for CO catalytic oxidation and sulfur resistance
Abstract
xMoO3-6CeO-Co3O4 catalysts with different Mo contents were prepared by coprecipitation method and impregnation method.CO conversion and sulfur tolerance properties of the catalysts were investigated.The catalysts were characterized by BET,SEM,FT-IR and H2-TPR techniques.The results indicated that the addition of MoO3 could improve the activity of the catalyst at low temperature.5.61MoO3-6CeO-Co3O4 catalyst possessed the best activities at low temperature,and CO conversion reached 98% at 40 ℃,While its performance for sulfur tolerance could last 90 min.
Keyword:
catalytic chemistry; CO catalysis; low temperature activity; sulfur resistance; MoO3
低温CO催化剂主要分为贵金属催化剂和非贵金属催化剂。研究较多的贵金属催化剂, 如Au、Pt和Pd催化剂具有催化活性高[1]和稳定性好, 但价格昂贵, 且在有水情况下易失活。非贵金属催化剂研究较为成熟的是Cu和Mn催化剂, 其中, Hopcalite催化剂为工业应用较多的CO催化剂。非贵金属催化剂价格低廉, 但耐硫和耐水性能较差。Co3O4具有较高的催化氧化性能[2], 尤其在低温环境下催化氧化性能更佳。Xie Xiaowei等[3]制备的纳米棒状Co3O4在-77 ℃对CO去除率达100 %。Wang Xi等[4]制备的Co/C-600在0 ℃和干燥气氛中, CO去除率达100%。CeO2的储氧能力较高, 与Co3O4结合, 可提高氧化还原性能和低温效率与稳定性。MoO3在脱硝反应中具有优异的抗硫性能[5]。
本文采用共沉淀法和浸渍法联用制备不同Mo质量分数的xMoO3-6CeO-Co3O4催化剂, 测试催化剂催化氧化CO效率及其耐硫性能, 并对催化剂进行BET、SEM、FT-IR和H2-TPR等表征。
1 实验部分
1.1 试剂与仪器
乙酸钴, 分析纯, 天津市福晨化学试剂厂; 硝酸铈, 分析纯, 天津市福晨化学试剂厂; 钼酸铵, 分析纯, 国药集团化学试剂有限公司; 无水乙醇溶液, 分析纯, 天津市福晨化学试剂厂; CO体积分数8%、O2体积分数99.9%, 工业纯、N2体积分数99.9%, 工业纯, 北京市海瑞通达气体有限公司。
傅里叶红外光谱分析仪, Thermo Fisher公司; ASAP 2050物理吸附仪, 美国麦克仪器公司; Autochem Ⅱ 2920型化学吸附仪, 美国麦克仪器公司; Hitachi S-4300扫描电子显微镜, 日本日立公司; KQ-2200E 型超声波清洗器, 昆山市超声仪器有限公司; JJ-1精密定时电动搅拌器, 江苏省金坛市荣华仪器制造有限公司; D07-7B型质量流量控制器、D08-1F型流量显示仪, 北京七星华创电子股份有限公司; CKW-1100型温度控制器, 北京市朝阳自动化仪表厂; testo 350烟气分析仪, 德国德图公司, 量程0~10 000× 10-6, 分辨率1× 10-6。
1.2 催化剂制备
采用共沉淀法和浸渍法联用制备MoO3-6CeO-Co3O4催化剂。先采用共沉淀法制备6CeO-Co3O4前驱体沉淀物, 对沉淀物进行洗涤去除钠离子以及乙酸根离子, 再采用浸渍法将Mo负载于6CeO-Co3O4前驱体上。110 ℃烘干, 400 ℃空气气氛焙烧, 研磨筛选(20~40)目颗粒物作为测试对象。
在一定量乙酸钴硝酸铈共沉淀后的沉淀物中分别添加0.2 g、0.4 g、0.6 g和0.8 g钼酸铵, 制备MoO3质量分数分别为1.92%、3.81%、5.61%和7.35%的MoO3-6CeO-Co3O4催化剂, 标记为xMoO3-6CeO-Co3O4(x表示Mo质量分数)。
1.3 催化剂评价
采用固定床评价装置, 反应管内径19 mm, 外径23 mm, 玻璃管壁厚2 mm。进口气体CO浓度1 200× 10-6, O2体积分数5%, 平衡气为N2, 总流量1. 5 L· min-1, 空速15 000 h-1, 测试温度(30~120) ℃。测试进出口CO浓度, 计算CO转化率。
1.4 催化剂表征
红外表征在傅里叶红外光谱分析仪上进行, 按一定比例105 ℃混合烘干4 h的KBr样品, 研磨后在压片机制成薄片, 对薄片进行红外测定。
物理吸附在ASAP2050物理吸附仪上进行, 比表面积由 BET 方程计算, 孔容和孔径由 BJH方程计算。对样品进行真空脱气, 90 ℃脱气1 h, 350 ℃脱气2 h, 升温速率10 ℃· min-1。
H2-TPR在AutochemⅡ 2920型化学吸附仪上进行, O2气氛400 ℃预处理30 min, 降至室温, Ar气吹扫60 min, 然后通入10%H2-Ar混合气, 升温速率10 ℃· min-1, TCD在线记录H2浓度。用CuO标样(纯度99.995%)标定H2消耗量。
催化剂表面形貌观察在扫描电子显微镜上进行, 工作电压15 kV。
2 结果与讨论
2.1 Mo含量对催化剂低温活性的影响
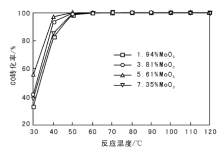
在不同反应温度下, MoO3含量对CO转化率的影响如图1所示。
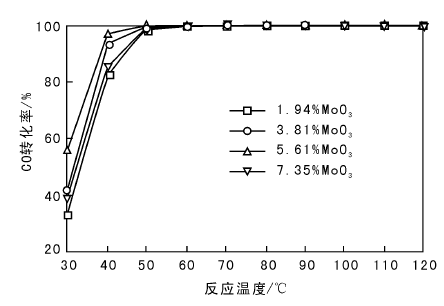 | 图 1 MoO3含量对CO转化率的影响Figure 1 Effects of different MoO3 loadings of the catalysts on CO conversion |
由图1可以看出, 添加质量分数5.61%MoO3的催化剂在30 ℃CO转化率达56%。MoO3添加量过大, 催化剂低温活性略降, 最优催化剂为5.61MoO3-6CeO-Co3O4。根据Langmuir-Hinswood机理, 反应时O2与CO吸附于催化剂表面, 在吸附状态下进行反应。随着温度升高, 催化剂对CO的解离能力逐渐提高。MoO3对CeO-Co3O4存在修饰作用, 改变了催化剂的电子结构和形态结构, 使氧化物之间的协同作用得以体现, 提高催化剂低温活性。
2.2 催化剂结构性能
考察MoO3含量对催化剂结构性能的影响, 结果如表1所示。
| 表 1 MoO3含量对催化剂比表面积、孔容和孔径的影响 Table 1 Effects of different MoO3 loadings of the catalysts on specific surface area, pore volume and pore size |
由表1可以看出, 随着MoO3质量分数增加, 催化剂比表面积和孔径先增后减, 比表面积最高为3.81MoO3-6CeO-Co3O4催化剂, 孔径最大为5.61MoO3-6CeO-Co3O4催化剂。孔容随着MoO3含量增加而增大, 最大为7.35MoO3-6CeO-Co3O4催化剂。
2.3 H2-TPR
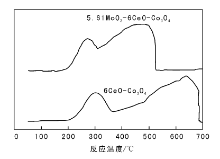
5.61MoO3-6CeO-Co3O4和6CeO-Co3O4催化剂的H2-TPR谱图如图2所示。
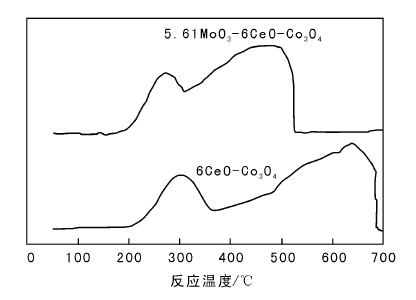 | 图 2 5.61MoO3-6CeO-Co3O4和6CeO-Co3O4催化剂的H2-TPR谱图Figure 2 H2-TPR profiles of 5.61MoO3-6CeO-Co3O4and 6CeO-Co3O4 catalysts |
由图2可以看出, 6CeO-Co3O4催化剂在约301 ℃和637 ℃出现两个还原峰, 添加MoO3后, 还原峰位置明显前移, 在256 ℃和443 ℃出现两个还
原峰。可能是MoO3掺杂会造成Co3O4结构缺陷增加, 加快氧在催化剂体相的流动性, 进而提高催化剂的低温活性。
2.4 耐硫性能
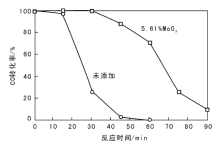
系统通入SO2气体, 考察5.61MoO3-6CeO-Co3O4催化剂的耐硫性能, 结果见图3。测试温度为T100, 空速15 000 h-1, SO2含量160× 10-6, CO含量1 200× 10-6, O2体积分数5%, 平衡气为N2。
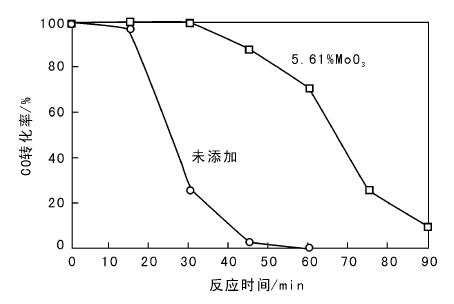 | 图 3 通入SO2后5.61MoO3-6CeO-Co3O4催化剂催化活性Figure 3 Effects of SO2 introduction on CO conversion over the 5.61MoO3-6CeO-Co3O4 catalysts |
由图3可以看出, 5.61MoO3-6CeO-Co3O4催化剂在30 min后CO转化率开始下降, 90 min后失活。未添加MoO3催化剂在15 min后CO转化率急剧下降, 45 min后失活。表明添加一定量的MoO3, 可增加催化剂的耐硫性能。
2.5 SEM
失活前后5.61MoO3-6CeO-Co3O4催化剂的SEM照片如图4所示。由图4可以看出, 新鲜催化剂表面有明显的孔状结构, 而失活催化剂表面不存在孔状结构。在较小的放大倍数下, 可以观察到催化剂表面形成的模糊团状物。可能是反应气体中SO2与催化剂表面的活性成分发生反应, 生成硫酸盐物质堵塞孔道, 降低催化性能。同时, 对失活前后催化剂进行BET表征, 结果表明, 催化剂比表面积由94 m2· g-1降至81 m2· g-1。
 | 图 4 失活前后5.61MoO3-6CeO-Co3O4催化剂的SEM照片Figure 4 SEM images of the fresh and deactivated 5.61MoO3-6CeO-Co3O4 catalysts |
2.6 FT-IR
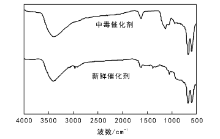
通入SO2前后的尾气通过傅里叶红外光谱进行定性定量测试, 结果表明, 催化剂失活前, 尾气仅存在CO2气体, 不存在CO和SO2。随着催化剂失活, 尾气中的CO和SO2逐渐增加, 至催化剂失活后CO和SO2含量与入口处相同。5.61MoO3-6CeO-Co3O4催化剂失活前后红外谱图如图5所示。
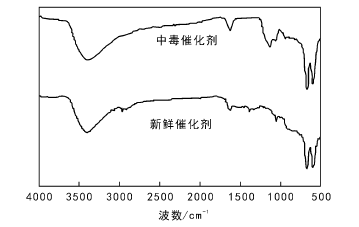 | 图 5 5.61MoO3-6CeO-Co3O4催化剂失活前后红外谱图Figure 5 FT-IR spectra of 5.61MoO3-6CeO-Co3O4catalysts before and after deactivation |
由图5可以看出, 660.81 cm-1和562.47 cm-1处出现Co— O键的伸缩振动吸收峰, 表明产物中含有Co3O4; 1 645 cm-1处出现Ce— O键的吸收峰, 表明产物中含有CeO2。中毒后催化剂在1 363 cm-1和1 383 cm-1处出现归属于典型的 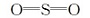
3 结 论
(1) MoO3的添加使6CeO-Co3O4催化剂上的Co3O4结构缺陷增加, 加快氧在体相中的流动性, 低温催化活性显著提高。其中, 5.61MoO3-6CeO-Co3O4催化剂低温活性最优, 40 ℃时CO去除率达97%。
(2) 添加一定量的MoO3可以使催化剂有一定耐硫性能, 但随时间延长, 孔道堵塞, 催化剂快速失活。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|