

{kind=link}
{kind=link}
催化剂制备与研究 中低温煤焦油加氢裂化催化剂的表征与分析
引用本文
黄晔, 樊安, 刘旭, 古道金, 雷雨辰, 李冬. 催化剂制备与研究 中低温煤焦油加氢裂化催化剂的表征与分析 [J]. 工业催化, 2017,25(8): 24-29.
Huang Ye, Fan An, Liu Xu, Gu Daojin, Lei Yuchen, Li Dong. Characterization and analysis of mid-low-temperature coal tar hydrocracking catalyst[J]. Industrial Catalysis, 2017,25(8): 24-29.
Doi:10.3969/j.issn.1008-1143.2017.08.005Huang Ye, Fan An, Liu Xu, Gu Daojin, Lei Yuchen, Li Dong. Characterization and analysis of mid-low-temperature coal tar hydrocracking catalyst[J]. Industrial Catalysis, 2017,25(8): 24-29.
Permissions
Copyright©2017, 《工业催化》编辑部
《工业催化》编辑部 所有
催化剂制备与研究 中低温煤焦油加氢裂化催化剂的表征与分析
作者简介:黄 晔,1971年生,男,陕西省神木县人,高级工程师,主要从事化工技术的开发及工业实践。
摘要
通过对反应前后的煤焦油加氢裂化催化剂进行表征与分析,研究影响中低温煤焦油加氢催化剂失活的原因,以煤焦油组分特点为依据,分别考察加氢裂化催化剂的活性元素、积炭、金属沉淀、分散度和中心酸性等对催化剂寿命的影响。结果表明,煤焦油加氢催化剂的失活原因主要为积炭失活、金属沉积和水热失活;催化剂中较低的金属钙含量、良好的水热稳定性及较低的L酸含量有利于催化剂寿命的提高。
关键词:
催化剂工程; 中低温煤焦油; 加氢裂化催化剂; 表面酸量
中图分类号:O643.36+3
文献标志码:A
文章编号:1008-1143(2017)08-0024-06
Characterization and analysis of mid-low-temperature coal tar hydrocracking catalyst
Abstract
The reasons for deactivation of hydrocracking catalyst for coal tar hydrotreating process in mid-low-temperature was studied by analysis the characterization of catalyst before and after the reaction.Based on the characteristics of coal tar,the influence of active elements,carbon deposition,metal deposition,dispersion and central acidity on the catalyst life was studied.The results showed that carbon deposition,metal deposition and hydrothermal deactivation were the main reasons for the deactivation of hydrocracking catalyst.Good hydrothermal stability,low content of Ca and Lewis acid in catalyst favored prolonging the hydrocracking catalyst life.
Keyword:
catalyst engineering; mid-low-temperature coal tar; hydrocracking catalyst; surface acidity
近年来, 随着石油资源日渐短缺, 煤化工相关产业获得重视, 而煤焦油加氢制备轻质燃料油项目不仅可以提高产品的经济价值又符合国家的能源策略, 鉴于中国“ 多煤、缺油、少气” 的能源结构[1], 要实现经济快速持续发展, 必须调整优化能源结构, 推动以煤为主的多元化发展。
加氢制备燃料油是目前对煤焦油最有效的利用方式, 其工艺核心是筛选/制备出合适的加氢催化剂。中低温煤焦油产地具有一定地域性, 并且油品较质劣。
催化剂失活是影响煤焦油加氢过程的关键因素, 而催化剂类型、原料油特性和加氢工艺条件等均可导致催化剂失活。Murray R Gray等[2, 3, 4]认为, 催化剂表面积炭是导致催化剂活性下降的最主要原因。Marafi A等[5]指出, 加氢脱金属催化剂应具有较好的金属包容能力, 该能力大小直接影响催化剂使用寿命。任杰[6]对催化裂化过程建立了水蒸汽分压与催化剂活性的动力学模型, 认为水蒸汽分压大小与催化剂活性成反比。Masuda T等[7]研究了高温下裂化催化剂的相变化, 认为催化剂失活是一个相态稳定化过程。白天忠等[8]研究发现, H2S对催化剂脱硫活性有明显抑制作用。Erley W等[9]认为, 烧结会改变催化剂微孔结构, 催化剂易产生相变化, 导致催化剂结构坍塌。
本文在固定床加氢反应器上, 使用两种不同类型催化剂, 对反应前后的煤焦油加氢裂化催化剂进行表征与分析, 研究影响中低温煤焦油加氢催化剂失活的主要原因及其机理。
1 实验部分
1.1 原料与催化剂
原料中低温煤焦油来自陕北某焦化企业, 热解温度为(893~1 023) K, 密度1.0372 g· cm-3, 50 ℃运动黏度14.22 mm2· s-1, ω (C)=7.62%, ω (H)=73.07%, ω (S)=0.35%, ω (N)=1.16%, ω (O)=17.8%。
采用的催化剂为自行研发的焦油加氢裂化催化剂A和B。
1.2 催化剂活性评价
加氢装置为北京某石化装备制造企业生产的200 mL双管式固定床三相加氢反应器。反应条件为:温度(663~683) K, 空速(0.2~0.4) h-1, 氢分压12 MPa, 氢油体积比1 200∶ 1, 催化剂A和B的总评价反应时间分别为1 500 h和2 500 h。催化剂A在反应末期, 活性明显下降, 产品油收率由初期的80%降至57%, 产品油中氮含量由30 μ g· g-1升高至200 μ g· g-1, 硫含量由小于10 μ g· g-1升至40 μ g· g-1; 催化剂B在整个评价过程中活性基本稳定, 产品油收率77%~81%, 产品油中氮含量为(30~50) μ g· g-1, 硫含量小于10 μ g· g-1。
将新鲜加氢裂化催化剂A、B分别标记为X-A和X-B; 失活加氢裂化催化剂用V(甲苯)∶ V(乙醇)=1∶ 1的混合溶剂进行抽提24 h至无色, 以除去样品上附着的可溶性油分, 抽提后催化剂A、B样品分别标记为S-A和S-B; 失活加氢裂化催化剂在15%氮气氛围以5 K· min-1的升温速率升温至873 K, 焙烧6 h, 以烧除催化剂表面积炭, 再生加氢裂化催化剂A、B样品分别标记为Z-A和Z-B。
1.3 催化剂表征
采用美国Thermo Scientific公司全谱直读等离子体发射光谱仪IRIS Advantage检测催化剂中的金属含量。
采用日本理学公司X射线衍射仪D/max-2400分析催化剂晶相, N2为载气, 扫描速率8° · min-1, 扫描范围3° ~90° 。
采用英国KROTOS公司光电子能谱仪800SIMS对催化剂的化学态进行分析, X射线源为铝Ka微聚集单色器, 离子枪能量为(100~4 000) eV, 可获取光斑大小为(30~400) μ m。
采用美国Nicolet公司红外吡啶吸附仪FTIR-500采集催化剂表面吡啶脱附图谱, 仪器扫描范围(400~4 000) cm-1, 分辨率4 cm-1, 扫描次数64次。
2 结果与讨论
2.1 催化剂宏观物性
不同状态加氢裂化催化剂的宏观物性见表1。
| 表1 不同状态催化剂的宏观物性 Table 1 Physical properties of catalysts in different states |
由表1可以看出, 失活加氢裂化催化剂A和B的比表面积和孔容均大幅下降, 表明催化剂在运转过程中产生大量积炭覆盖在表面, 堵塞了催化剂表面部分微孔, 造成卸出的催化剂宏观物性降低。
再生加氢裂化催化剂的比表面积、孔容和平均孔径均有恢复, 再生加氢裂化催化剂A的比表面积和孔容恢复率低于再生加氢裂化催化剂B, 可能是反应后期再生加氢裂化催化剂A活性下降, 原料中部分金属离子沉积在催化剂表面, 这类金属离子经过焙烧后, 部分迁移进入催化剂孔道造成再生催化剂宏观物性恢复率下降。再生加氢裂化催化剂A平均孔径大幅提高, 表明高温下迁移入催化剂孔道内的金属发生熔结, 进一步撑大催化剂孔道。
失活加氢裂化催化剂A和B的径向抗压碎力均下降, 失活加氢裂化催化剂B的径向抗压碎力降至约为新鲜催化剂的50%, 这可能是由于新鲜催化剂B的水热稳定性不佳, 在反应管内与加氢反应产生的蒸汽长期接触, 导致卸出催化剂的径向抗压碎力大幅降低。
总体看来, 催化剂B在整个评价阶段始终保持较高的反应活性, 卸出的催化剂样品未发现有明显破碎现象, 且失活和再生加氢裂化催化剂的孔径和比表面积均未发生大幅降低。水热失活在较高反应温度[(1 005~1 116) K]下, 成为催化剂失活主因[10]。因此, 失活和再生加氢裂化催化剂B的径向抗压碎力下降幅度较大可能仅与其自身性质有关, 在催化剂设计时需要提高催化剂自身的水热稳定性, 正常操作下, 反应器内生成的大量水汽不会对催化剂稳定性造成显著影响。
2.2 金属元素含量
新鲜和再生加氢裂化催化剂A和B的金属含量检测结果见表2。加氢裂化催化剂A、B均为Ni、Mo和W活性元素为主的负载型加氢裂化催化剂, 并含有微量的助剂金属元素如Na、Mg、Ca和Fe等。
| 表 2 新鲜和再生加氢裂化催化剂A和B的ICP检测结果 Table 2 ICP detection results of fresh and regeneration hydrocracking catalysts A and B |
从表2可以看出, 再生加氢裂化催化剂A中的Ni和W损失率分别为1.66%和1.42%, 再生加氢裂化催化剂B中的Ni和W损失率分别为1.71%和1.73%。由于高温热作用, 催化剂在长期运行中部分活性组挥发或活性金属烧结, 造成金属分散性变差, 这是造成少量金属流失的原因, 但活性金属元素流失不是造成催化剂失活的主要原因。
再生加氢裂化催化剂表面上的Ca元素含量明显增加, 而其余金属元素含量变化不大。表明:(1) 煤焦油中较少的Ni和V元素在前期加氢脱金属过程就可以除去, 因此, 对加氢裂化催化剂几乎没有影响; (2) Fe、Mg和Na等元素在高温、高压和硫化氢存在条件下, 迅速反应生成相应的金属硫化盐, 因此, 该类金属的沉积主要发生在反应器顶部(如保护剂, 缓冲剂), 而反应器末端的沉积量较少。(3) 研究发现, Ca元素在整套加氢催化剂中均有分布且分布量呈依次递减趋势, 这是由于煤焦油中Ca主要以酚钙和环烷酸钙等有机盐形式存在, 该类Ca盐是一种极性较强的配合物, 容易与煤焦油中的极性较强的胶质和沥青质吸附在加氢催化剂表面, 并在活性金属作用下逐渐发生加氢脱钙反应生成CaS的沉淀, 将催化剂表面包覆, 与文献[11]相吻合。
2.3 XRD
图1为不同状态加氢裂化催化剂A和B的XRD图。从图1可以看出, 新鲜加氢裂化催化剂在2θ =14.4° 存在较强的衍射峰(归属于活性金属W), 催化剂硫化后归属于WS2晶体的Bragg衍射峰。再生加氢裂化催化剂在14.4° 的衍射峰几乎不存在, 因此, 硫化后活性金属W在催化剂表面始终保持高度分散。再生加氢裂化催化剂在2θ =26.6° 存在明显的衍射峰(归属于β -NiMoO4), 表明活性金属Ni存在聚集, 有效金属活性相减少。加氢裂化催化剂A、B的载体均为γ -Al2O3, 催化剂A载体的晶型结构未出现明显变化; 而催化剂B的载体出现明显的聚集现象, 载体的晶型向更为稳定的α -Al2O3发生转变, 这一过程会造成催化剂孔道坍塌, 比表面积下降。造成催化剂晶型转化的原因可能是催化剂载体的自身缺陷和较差的热稳定性。
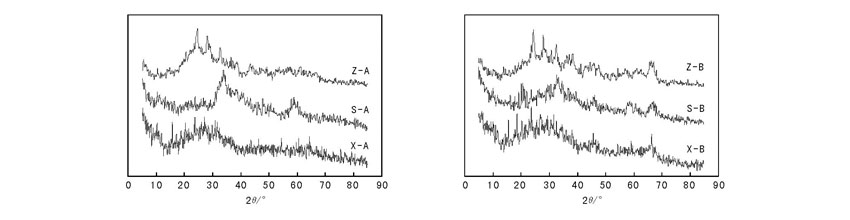 | 图 1 不同状态加氢裂化催化剂A和B的XRD图Figure 1 XRD patterns of hydrocracking catalyst A and B in different state |
2.4 C1s XPS
失活加氢裂化催化剂A和B的C1s XPS谱图及其分峰拟合结果图2和表3。从表3可以看出, 失活加氢裂化催化剂A和B的石墨化程度相近, 而失活加氢裂化催化剂A的C— C化合物物质的量分数(21.1%)远大于失活加氢裂化催化剂B(13.1%), 这是由于在反应末期, 催化剂A的表面生成大量堵塞活性中心的“ 新生成焦” , 催化剂活性迅速下降。由于催化剂是快速失活, 其表面石墨化程度不是很高, “ 新生成焦” 的产生是造成催化剂失活的主要原因。另外, 失活加氢裂化催化剂A的C— OR化合物物质的量分数(25.8%)小于失活加氢裂化催化剂B(31.5%), 表明C— C化合物可能是由C— OR脱水缩合形成。
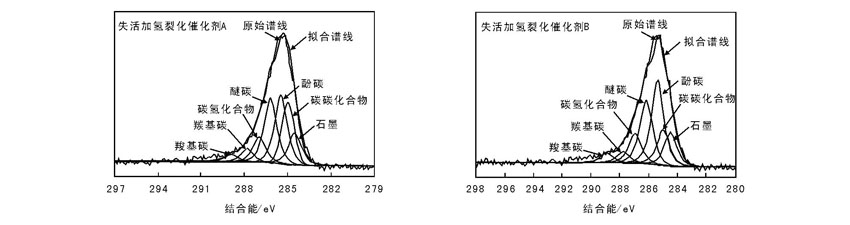 | 图 2 失活加氢裂化催化剂A和B的C1s XPS谱图Figure 2 C1s XPS spectra of the deactivation hydrocracking catalysts A and B |
| 表 3 失活加氢裂化催化剂A和B的C1s XPS分峰拟合结果 Table 3 Fitting results of C1s XPS peak of the deactivation hydrocracking catalysts A and B |
2.5 催化剂表面酸量
不同状态加氢裂化催化剂表面酸量检测结果见表4。由表4可以看出, 新鲜加氢裂化催化剂A、B中B酸含量明显小于L酸含量, 表明催化剂上能够接受电子对的空穴较少, 对于氮含量较高的原料(如煤焦油、VGO), 氮存在形式主要为吡啶等碱性氮, 而相较于B酸, 碱性氮更容易在L酸中心上产生积炭[12], 因此, 较低的B酸含量可以有效抑制碱性氮的积炭沉淀。
失活加氢裂化催化剂A的L酸量损失率为22.8%, 远大于失活加氢裂化催化剂B, B酸量损失率相差不大, 表明原料中的碱性氮与催化剂的酸性相中心接触(主要是L酸中心), 由于催化剂活性下降, N原子无法有效脱除, 碱性氮则吸附在催化剂的活性中心, 形成较强的化学吸附键, 加深了积炭产生并将酸性中心深深包埋, 降低催化剂中心酸度。
再生加氢裂化催化剂A的中心酸度大幅恢复, L酸量恢复至新鲜催化剂的88.6%, 表明烧炭可以除去原本阻塞孔道的碱性氮和积炭, 而催化剂自身酸性未出现较大损失。再生加氢裂化催化剂B中L酸量和B酸量大幅度降低, 根据对催化剂宏观物性和ICP的分析结果, 催化剂酸量降低是由于催化剂强度下降, 热稳定性变差, 高温烧炭造成催化剂原因孔道崩塌, 这是一种不可逆的酸量下降过程。
| 表 4 不同状态加氢裂化催化剂表面酸量检测结果 Table 4 Test results of surface acidity of hydrocracking catalysts in different states |
3 结 论
(1) 通过对反应前后的煤焦油加氢裂化催化剂进行表征与分析, 发现导致加氢裂化催化剂A失活的原因主要是催化剂积炭和活性组分流失; 加氢裂化催化剂B载体存在明显的裂纹, 水热稳定性较差, 可能是由于载体自身存在缺陷所致。
(2) 原料中的金属Ca元素对催化剂影响较大, Ca沉积在催化剂表面形成积垢, 部分Ca元素迁移进入催化剂微孔, 发生熔结, 使孔径增大。
(3) 煤焦油原料中含有大量酚, 加氢反应后转化为水蒸汽存在于反应器内, 热稳定性较差的催化剂载体在高温和高水汽分压下容易发生热崩, 因此, 煤焦油加氢催化剂要求载体具有良好的热稳定性。
(4) 煤焦油原料中的碱性氮主要吸附在加氢裂化催化剂的L酸酸性中心上并能促进积炭的产生。催化剂经过再生后, 原本阻塞孔道的碱性氮和积炭被除去, 酸性得到大幅恢复。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|