{kind=link}
{kind=link}
{kind=link}
稠环芳烃裂化研究进展
[孙书桩, 彭鹏, 阎子峰*  ]
]
]
|
|


作者简介:孙书桩,1995年生,男,在读硕士研究生,研究方向为催化材料与催化剂。
稠环芳烃物种含有大量的芳烃资源,由于苯环间的共轭效应,难以将其直接转化。随着原油芳烃含量的逐年增高,在工业规模如何有效地处理稠环芳烃备受关注。对稠环芳烃的加氢裂化和催化裂化反应机理进行总结对比,并通过介绍裂化催化剂的研究进展为稠环芳烃的催化裂化拓展新思路。
Polycyclic aromatic hydrocarbons contain large amounts of aromatic hydrocarbons,which are difficult to convert directly due to the conjugated effect of benzene rings.In recent years,the content of aromatics in crude oil has been increasing year by year.How to crack polycyclic aromatic hydrocarbons on an industrial scale has attracted much more attention.The mechanism of hydrocracking and catalytic cracking of polycyclic aromatic hydrocarbons have been summarized and compared.By introducing the progress of corresponding catalysts,new ideas might be developed for the catalytic cracking of polycyclic aromatic hydrocarbons.
随着石油资源的开采, 全球范围原油的重质化趋势日益明显, 重质油中不仅含有大量的沥青质和胶质, 还含有大量的稠环芳烃。由于苯环的共轭效应, 这类物质化学性质十分稳定, 且稠环芳烃是一种不容忽视的环境污染物, 部分种类的稠环芳烃还有较强的生物毒性, 有些稠环芳烃是已知的致癌物质和诱变剂, 稠环芳烃已被列入美国环保署的优先污染物清单[1]。当前, 沥青质的应用主要集中在铺路和涂料等领域, 不仅对环境造成极大污染, 而且也浪费了大量的芳烃资源, 如果通过催化剂的引入能从稠环芳烃中裂化出高附加值的低碳化合物和芳烃及其衍生物, 将成为石化垃圾高效利用的重要途径。
本文从稠环芳烃的裂化反应机理和裂化催化剂进展等方面综述稠环芳烃裂化的研究进展。
加氢裂化能将稠环芳烃通过加氢、裂化和异构化等反应转化为轻质芳烃和链烃等, 既可有效地脱除稠环芳烃, 又可提高油品十六烷值, 稠环芳烃的加氢裂化需要至少饱和一个苯环, 如萘至少要转化为四氢萘才能够进行裂化, 且四氢萘等可作为煤液化反应的供氢溶剂, 十氢萘、四氢芘也是重要的功能材料和医药中间体[2]。稠环芳烃加氢裂化反应一般包括加氢、异构化、氢转移、开环裂化及脱氢缩合生焦反应, 反应机理的研究主要在萘、蒽和菲等模型化合物上开展, 加氢裂化反应实质上就是催化裂化碳正离子反应伴随加氢反应[3]。
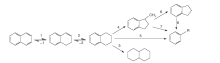
以结构最简单的稠环芳烃萘为例, 萘的加氢裂化网络如图1所示。萘的一个苯环首先发生加氢反应, 直到成为饱和烷烃环方可进行下一步开环、异构化或者进一步饱和的过程。萘的加氢和裂化普遍被认为遵循碳正离子机理, 萘可被催化剂上的B酸催化形成质子化的萘[4], 质子化的萘可以与一个H-形成二氢萘, 或者与另一个萘分子通过Scholl反应形成二氢联萘[5]。
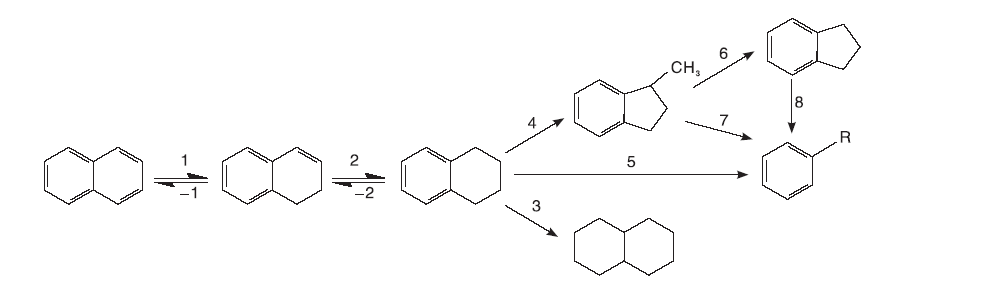 | 图1 萘的加氢裂化反应网络Figure 1 Hydrocracking reaction network of naphthalene |
加氢处理较大稠环芳烃分子的过程相对复杂, Korre S C等[6]发现, 加氢按环逐一进行, 随环数的增加, 加氢反应性上升, 吸附系数增大, 若环数相同则加氢反应性随烷基或环烷数增加而上升, 端环加氢快于中环。
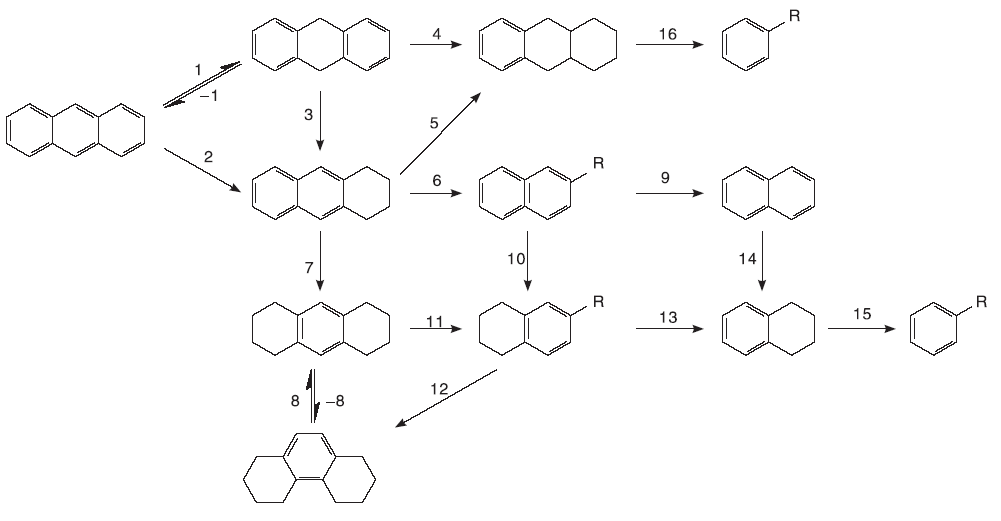
稠环芳烃分子由于其极强的苯环共轭作用, 化学性质十分稳定, 由于苯环极难裂化, 直接催化裂化稠环芳烃难度极大, 可能的稠环芳烃催化裂化需要经过3个步骤, 即稠环芳烃的缩合(氢源)、稠环芳烃的加氢(氢转移)和稠环芳烃的裂化。虽然催化裂化稠环芳烃分子需要牺牲更多的稠环芳烃分子来供氢, 但是由于该路径无需任何外部供氢, 成本较稠环芳烃加氢裂化低, 且稠环芳烃在当前炼油工业中难以实现转化, 多以废料废渣形式存在, 稠环芳烃的催化裂化既可以在低成本下变废为宝, 也避免了大量的稠环芳烃排放到环境中。若能实现对稠环芳烃非氢气氛下的裂化, 将解决石油工业中大量的稠环芳烃, 并将这部分难以处理的工业废料变废为宝, 降低其环境排放等。
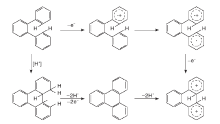
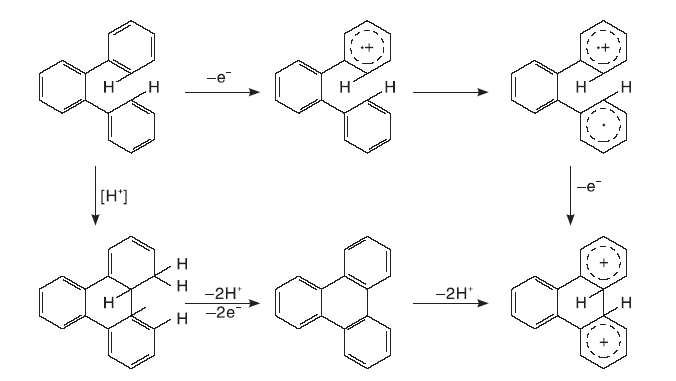
稠环芳烃的生长为两个或两个以上小稠环芳烃缩合成更大稠环芳烃的过程。在1910年, Scholl R完成了以三氯化铝为催化剂用萘合成醌[7]和二萘嵌苯[8]的工作, 自此研究者将这类反应统称为Scholl反应或Scholl凝结, 即借助L酸和B酸在两种芳烃化合物之间的偶联反应[9]。Benjamin认为, Scholl反应中的B酸通常是L酸的杂质, 也在Scholl反应过程中形成[10], 其使用的催化剂大多为金属氯化物如氯化铁、氯化铝和氯化钼等。关于B酸和L酸在Scholl凝结中分别起到的作用众说纷纭, 不过由L酸部分转化得到的B酸参与到该反应中得到较多认可。Scholl反应最主要的特征就是在两个苯环之间生成C— C键, 生成分子间的C— C键需要较高的温度和酸性较强的催化剂, 分子内的这一过程更易发生。
Scholl反应的机理目前还没有统一认识, 从已有的工作看, 特定的金属离子(如Al3+和Mo5+)能起到较好的催化效果。Scholl反应机理推测主要有自由基机理和碳正离子机理。自由基机理第一步是通过氧化将一个反应物分子转化为自由基阳离子, 形成的自由基阳离子与第二个反应物分子发生取代反应, 并且新的自由基离子形成带有正电荷的一个环和另一个带有自由基的环, 第三步将二氢化合物分解成联芳基化合物。在碳正离子机理中, 一个反应物分子被质子化为一个离子, 之后攻击第二个反应物分子。金属离子也可以通过L酸的侵蚀形成。这些机制难以区分, 因为许多L酸可以表现为氧化剂, 在室温下与已知的单电子氧化剂发生的反应可能通过自由基阳离子机理进行, 需要升高温度的反应则可能通过碳正离子机制进行[11]。
分子间的氢转移是稠环芳烃催化裂化关键所在, 通过Scholl反应而缩合的稠环芳烃产生的少量氢便是稠环芳烃能否催化裂化的核心点。Salim S S等[12, 13]研究了常见的双环和三环稠环芳烃在金属氯化物催化作用下的反应, 重点比较了氯化锌和氯化铝对稠环芳烃的催化效果, 通过实验设计考察了稠环芳烃分子反应结果中饱和环中氢的来源。分别以氯化锌和氯化铝为催化剂, 在相同压力的氢气氛和氮气氛下催化稠环芳烃反应, 当气氛由氢气气氛转变为氮气气氛后, 以氯化锌为催化剂的蒽和1-萘酚转化率分别从68%和25%下降到< 1%和4%, 相比起来, 氯化铝为催化剂反应物分子的转化率和产物组成变化均小得多。这一现象表明, 氯化铝作为催化剂时, 反应过程的脱氢加氢均是在反应物分子之间完成, 即使没有氢分子的引入, 也能较高效地发生氢转移反应。
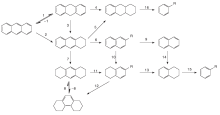
稠环芳烃的催化裂化反应实质是部分稠环芳烃缩合成更大分子量的稠环芳烃甚至成为焦炭, 这一过程产生的一定量的氢可作为氢源供给小分子量的稠环芳烃加氢裂化, 宝贵的氢源用在何处以及接受氢源的稠环芳烃分子加氢到什么程度就显得十分重要。Salim S S[12, 13]通过分析比较氮气气氛和氢气气氛下的产物含量, 大致得出图3中主要反应的优先级, 蒽转化为二氢蒽要比转化为四氢蒽迅速, 但是二氢蒽转化为四氢蒽速率要慢于蒽转化为四氢蒽, 由二氢蒽进一步加氢生成四氢蒽的难度远远小于生成六氢蒽的难度, 对称型八氢蒽的生成难度远小于非对称型八氢蒽。如图3中步骤1→ 3→ 7难度较步骤1→ 4小, 由四氢蒽生成非对称八氢蒽的难度小于二氢蒽生成非对称八氢蒽的难度, 如图3中步骤5发生难度小于步骤4。
双环稠环芳烃萘的加氢过程与蒽不同, 萘首先加氢得到二氢化萘, 但是二氢化萘反应活性很高, 会很快转化成四氢萘或者萘, 进一步加氢或者裂化异构化均通过四氢萘进行。
Scholl反应进行的程度是由催化剂的酸性和反应物的碱性决定, L酸转化成B酸(如氯化铝)的酸强度明显强于弱L酸的氯化锌[14], 所以以氯化铝为催化剂稠环芳烃更易于发生氢转移和Scholl反应。形成二氢萘的氢源仅有两种可能, 一是从别的碳氢物种中, 其实是来源于Scholl反应[15]; 二是外部供氢。当稠环芳烃分子的一个或多个苯环饱和后, 烷烃环的开环和异构化便可顺利进行, 最终达到处理稠环芳烃的效果。
裂化在炼油工业中比例极大, 是将原油中大分子裁剪成低碳烷烃、烯烃和汽柴油等组分的重要过程, 依据其裂化原理可分为加氢裂化和催化裂化, 两者均可有效处理稠环芳烃, 而具体的活性组分和裂化效果各有不同。
稠环芳烃的加氢裂化即用外部氢源将稠环芳烃部分或全部的芳环饱和后将烷烃环裂化的过程。稠环芳烃的加氢裂化过程中, 多芳环的部分氢化和所得环烷环的裂解是加氢裂化反应的关键步骤, 因此, 金属负载于酸性催化剂上的双功能催化剂被大量应用[16]。大多数研究者采用过渡金属或贵金属催化剂用于加氢裂化, 如负载于SiO2-Al2O3或沸石上的Mo、W、Co-Mo、Ni-Mo、Ni-W、Pt或Pt-Pd金属[17, 18], 这些种类的复合催化剂在较高的反应温度(7731 073) K和较高的氢压(815) MPa条件下, 苯、甲苯和二甲苯(BTX)收率仍低于50%。之后, 有研究者发现过渡金属的磷化物有较好的催化效果, 在多种的过渡金属磷化物中Ni2P有最优的加氢裂化活性[19, 20]。Kim Y S等[21]研究发现, Ni2P/SiO2催化剂在3.0 MPa和673 K条件下, 仅能将萘转化为四氢化萘和十氢化萘, 而负载于分子筛上的Ni2P的加氢裂化活性为Ni2P/Beta> Ni2P/USY> Ni2P/ZSM-5, Ni2P/Beta的BTX产率最高可达94.4%, 归因于其独特的中等酸度和孔隙度与良好分散的Ni2P相的氢化活性, 且Ni2P/Beta在反应过程中能保持局部和体积结构稳定。
稠环芳烃加氢裂化催化剂通常为双功能催化剂, 由活性金属和酸性中心组成。加氢活性组分主要由贵金属和非贵金属构成。侯冬梅等[22]研究表明, Pd/Al-MCM-41催化剂具有较高的脱硫活性和异构化活性, Pd/MY(Si-MCM-41-HY)催化剂表现出较高的加氢裂化活性。Jacquin M等[23]等研究发现, 将两种金属(如Pd和Pt)复配使用, 可以提高催化剂的加氢性能。而常用的非贵金属主要有Ni、Mo和W等。Haynes Jr H W等[24]以萘、蒽、菲和芘等结构较为简单的稠环芳烃分子作为模型化合物, 使用负载Ni-W的USY对稠环芳烃进行加氢裂化, 并取得不错的效果, 且USY在加氢裂解芘(临界直径0.9 nm)和较小的分子时非常活泼。Fu W等[25]研究发现, NiMoS/MZSM-5催化剂上菲基本完全转化, 得到的深度加氢产物八氢菲选择性达80.7%, 全氢菲选择性为17.9%。碳化物、氮化物和磷化物具有类贵金属的性质, 且成本低廉, 也可作为贵金属催化剂的潜在替代材料。
稠环芳烃的催化裂化过程比较复杂, 作为稠环芳烃催化裂化起点的Scholl反应使用的催化剂以金属氯化物和氟化物为主, 主要起催化作用的有强L酸位和B酸位。Salim S S等[12, 13]认为, 必须有B酸存在的催化剂才对萘和蒽的系列反应有较好的催化效果, 而B酸位则是通过L酸位与氯化氢、水或者乙醇作用得到; 并通过实验发现, 无水的氯化锌和氯化铝在无水环境下基本没有催化效果, 也直接支持了上述推测。有观点认为, 强酸性位尤其是L酸位和氧化剂是Scholl反应所必须的[26], 有些试剂可以同时起到两个作用, 而B酸位则是由L酸位转化且作为Scholl反应一定会生成的副产品出现, B酸位的存在同时加速了Scholl反应[17]。
稠环芳烃大量存在于渣油和沥青质中, 与之相伴的还有许多烷烃大分子, 催化裂化在处理重油大分子中扮演着十分重要的角色, 在当前的炼油工业中, 固体酸催化剂占据主导地位。分子筛是一种重要的多孔材料, 因其具有较高的比表面积、灵活多变的骨架结构、较好的水热稳定性、优异的离子交换性以及成本低廉等特点, 在石油炼制、石油化工和污水处理等领域发挥着巨大作用[27]。分子筛作为一种固体酸催化剂, 其酸强度和酸密度等相关性质可以通过调节硅铝比、同晶取代和适当的酸碱处理进行调控, 其孔结构也可以通过相应的酸碱处理或调节合成条件来调整, 进而提升其催化性能。
关于稠环芳烃的研究工作主要集中在加氢裂化和Scholl反应, 加氢裂化由于耗氢量巨大, 虽有较好的裂化效果, 却不具有实际工业应用价值。稠环芳烃的催化裂化将会是一个低投入高回报的处理方法, 虽然直接使用分子筛作为稠环芳烃催化裂化催化剂的工作较少, 但B酸位和L酸位对Scholl反应和稠环芳烃加氢裂化的效果均有许多工作支持, 且Scholl反应可以为稠环芳烃的加氢提供氢源。
分子筛催化剂是一种含有B酸和L酸酸位的固体酸催化剂, 可以通过一系列的改性方法调节分子筛的酸强度和酸密度。考虑到Scholl反应大量的稠环芳烃聚合以及研究需要的氢转移和饱和环的裂化, 再根据相关分子的分子动力学直径, 稠环芳烃的加氢和裂化在分子筛内部孔道进行, 稠环芳烃的聚合甚至生焦在分子筛外部进行会是一种解决方案。由相关分子的动力学直径以及这些分子分别在MFI孔道和FAU孔道内的扩散能垒差距, FAU结构更有利于体系内稠环芳烃分子的扩散[28]。稠环芳烃的催化裂化体系发生的是分子间的氢转移, 酸密度的调控或将对反应产生影响。本体系的氢来源于Scholl反应以及生焦的过程, 本身氢源处于稀缺状态, 控制本就稀缺的氢源在孔道深处参与稠环芳烃的加氢与裂化不现实, 较好的反应位置应该在孔口处。Y型分子筛为微孔分子筛, 其十二元环孔口直径为0.74 nm, 由于具有开放的孔道结构, 使多种有机分子能够进入孔道并且发生催化反应[29], 但是未经任何处理的Y型分子筛由于较大直径的分子在分子筛微孔孔道内存在, 导致较大的传质阻力, 并且易积炭失活, 严重影响催化剂的使用寿命[30]。尽量避免孔内生焦, 一是孔内生焦会严重堵塞催化剂孔道, 阻碍反应物分子和产物分子的扩散; 二是孔内生焦会迅速覆盖活性位点, 导致催化剂失活。
传统裂化催化剂可除去侧链(> 乙基)和环烷烃环, 留下相对较少的(仅13个甲基和乙基取代基)芳族核, 主要为萘、菲、芘、䓛和更重的组分。因此, 这些较小的芳族分子成为在反应器区域中焦炭形成反应的重要参与者, 且这类组分广泛存在于裂化过程中的循环油中[31]。生焦过程对传统催化体系不利, 对稠环芳烃的催化裂化甚之。稠环芳烃的催化裂化不可避免要大量生焦以提供稠环芳烃加氢裂化所必须的氢源, 所以控制生焦使其对催化过程的影响越小越好。
对分子筛的改性应用已有多年历史, 可以通过相应的酸碱处理和负载不同的金属等方法调节分子筛的孔结构和酸性质。稀土负载的分子筛催化剂不仅有着较好的水热稳定性, 且稀土离子在分子筛笼内通过极化和诱导作用增加了骨架硅羟基和铝羟基上电子向笼内的迁移概率, 增大了分子筛笼内的电子云密度, 使羟基表现出更强的酸性, B酸强度增加, 也相应提高了催化剂活性[32]。有文献对稀土改性 Y 型分子筛的酸性进行研究, 发现改性Y型分子筛的酸密度、总B酸量和强B酸量、总L酸量和强L酸量均有所增加; 当稀土含量达到某一峰值后, 酸密度、B酸量和L酸量开始下降。四配位、五配位和六配位的铝原子会影响B酸产生, 低配位的非骨架铝原子则会影响L酸的产生[33]。非骨架铝的存在在稠环芳烃的催化裂化中或许有其特别的效果, Hosseinpour N 等[34]研究了Y型分子筛和无定形二氧化硅-氧化铝的协同作用及对积炭的影响, 发现无定形二氧化硅-氧化铝包覆在Y型分子筛上, 避免活性中心直接暴露在残渣中, 催化活性提高, 有效避免了焦炭在活性位点上的生成(牺牲无定形二氧化硅-氧化铝用于生焦)。这些无定型二氧化硅-氧化铝不是与活性分子筛催化剂简单的机械混合, 作为包覆在分子筛表面用于焦炭生长的附着点, 这或许对稠环芳烃催化裂化的实现有借鉴意义。
近年来, 随着原油品质的下降, 重油的开采利用量日益升高, 其中大量难以处理的稠环芳烃是阻碍石化行业发展的一块绊脚石, 虽然研究者已经开发出多种处理稠环芳烃的方法如加氢裂化, 但均受限于高额的成本和难以实现工业化等等。Y分子筛广泛应用于石油加工、石油化工以及精细化工等各个领域, 尤其在催化裂化领域更是作为催化剂的主活性组分, 在深入理解稠环芳烃的催化裂化机理前提下, 对传统催化裂化催化剂进行有方向的二次设计, 使传统催化裂化催化剂能够控制稠环芳烃间氢转移以达到预期的效果, 赋予传统催化裂化工艺处理稠环芳烃的能力, 是原油重质化和劣质化大环境下石化行业进一步良好发展的契机。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|