


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
NiO(111)表面重构对乙烷C—H键活化影响的DFT+U研究
[喻婷婷1  , 宋卫余
, 宋卫余1 , 赵震1, 2, * , 刘坚1 ]
, 宋卫余, 刘坚|
|


作者简介:喻婷婷,1993年生,女,在读硕士研究生,主要从事乙烷氧化脱氢的密度泛函理论研究。E-mail:tingtingyu_cup@126.com
采用密度泛函理论(DFT)和Hubbard U值校正方法研究NiO(111)表面重构及其对乙烷C—H键断裂的影响。通过电子局域函数(Electron Localization Function,ELF)、态密度(Density of states,DOS)、过渡态结构和Bader charge等对表面重构和乙烷C—H键活化进行分析。结果表明,重构表面相比于理想表面更加稳定,同时重构表面上C—H键的活化更加困难。
Effects of surface reconstruction of NiO(111) facet on cleavage of C—H bond in ethane were investigated by density functional theory(DFT) and Hubbard U value correction methods.Surface reconstruction and ethane C—H bond activation were analyzed by electron localization function(ELF),density of states(DOS),transition state structure,and Bader charge.The results showed that the reconstructed surface was more stable than the ideal surface and the activation of C—H bond on the reconstructed surface was more difficult.
乙烯是石油化工行业的重要基石, 传统制乙烯采用石脑油或乙烷蒸汽裂解法, 制备过程温度高(大于800 ℃)[1], 能源消耗大, 且产物容易深度氧化为COx, 乙烯产率较低。催化氧化乙烷脱氢制乙烯不仅能降低反应温度, 提高乙烯选择性, 还能有效降低乙烯的生产成本。常用于乙烷氧化脱氢反应的催化剂有Ⅴ 基[2, 3, 4]、Mo基[5, 6]和Ni基[7, 8, 9]催化剂, 其中, Ni基催化剂以其优越的低温氧化性能被广泛研究。普遍认为, 在NiO上乙烷的氧化脱氢遵循Mars-van Krevelen机理, 乙烷C— H键的活化是整个反应的决速步骤[10]。Fung V等[11]运用DFT+U方法研究了Co3O4(111)面上乙烷的氧化脱氢反应, 计算结果显示, 乙烷C— H键断裂存在均裂和异裂两种方式, 均裂有利于第一个C— H键断裂, 而第二个C— H键断裂通过异裂方式能垒较低。文献[12, 13, 14, 15, 16]研究了烷烃分子在IrO2(110)和PdO(101)面上的C— H键活化和氧化脱氢反应。结果表明, 烷烃分子通过与表面金属原子形成σ 复合物, 使C— H键拉长, 从而促进C— H键断裂, 断裂后的H吸附到配位不饱和O原子上形成羟基, 烷基分子与金属原子结合。NiO是典型的p-型半导体, 表面容易形成阴离子和阳离子空位[17]。Varghese J J等[18]研究了NiO(100)和NiO(110)表面甲烷活化及空位对甲烷活化的影响, 研究发现, NiO(110)表面的活性优于NiO(100), O空位对甲烷的活化没有太大影响, 但Ni空位能影响空位附近原子的电子性质, 从而降低C— H键断裂的能垒。NiO为反铁磁(AFM)Ⅱ 型固体[19], NiO(111)表面是由带负电的氧离子层和带正电的镍离子层交替排列, NiO(111)表面性质较为特殊, 且稳定性较差, 在反应过程中极易发生重构现象。本文进行NiO(111)表面重构对乙烷C— H键活化影响的DFT+U研究。
计算均基于密度泛函理论[20]和赝势平面波基组的VASP[21](Vienna ab initio simulation program)程序, 电子交换关联势采用广义梯度近似法(generalized gradient approximation, GGA)中的Perdew-Burke-Ernzerh(PBE)泛函[22]。由于Ni含有3d(3d84s2)电子, 而DFT方法无法准确描述NiO的电子性质, 所以对Ni加Hubbard U值[23]校正, 有效的U-J值取5.3 eV, J=1 eV[18]。周期性的NiO(111)层板模型采用4× 4超胞, 6层原子, 其中底部两层原子固定, 真空层1.5 nm。计算参数平面波阶段能400 eV, 原子力的收敛判据10-4 eV, 布里渊区仅选取Gamma点[1]。乙烷C— H键断裂过渡态采用NEB[24](nudged elastic band)方法搜索, 每个过渡态通过有且仅有一个的虚频来验证。乙烷等小分子的优化均在1.0 nm的立方胞中进行。
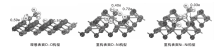
NiO(111)理想表面为极性表面, 垂直表面方向存在偶极矩, 且随表面厚度增加, 表面极性增大, 表面稳定性逐渐降低, 最后发生重构现象, 使表面重新回到稳定状态。NiO(111)重构表面有多种不同构型, 如alpha结构、vacancy结构和octopolar结构等, Wolf D[26]提出的p(2× 2)octopolar结构被认为是表面发生重构以后形成的最稳定结构, 如图1所示。重构后每个O原子分别与3个Ni原子成键, 形成类似三角锥构型。由于NiO(111)表面为Ni原子层和O原子层交替排列, 所以可以用Ni— O键长代替层间距。
 | 图1 模型结构图 1代表O原子; 2和3分别代表自旋向下和自旋向上的的Ni原子Figure 1 model structures |
与体相NiO相比, Ni(111)理想表面和重构表面Ni— O键长发生改变。表1为NiO(111)理想表面与重构表面Ni— O键键长对比。
| 表1 NiO(111)理想表面与重构表面Ni— O键长对比 Table 1 Comparison of Ni— O bond length between ideal and reconstructed surface of NiO(111) |
由表1可见, 体相Ni— O键长为0.208 9 nm, 理想表面结构优化后, 表层O原子向内弛豫, 第二层Ni原子向外弛豫, 造成层间距缩小, Ni— O键长减小到0.191 8 nm。这是因为体相的Ni和O原子均是六配位, 而形成NiO(111)表面后, 表面原子配位数减小, 表面存在不稳定的悬挂键, 为使表面达到稳定状态, 原子会通过缩短彼此间的键长克服悬挂键造成的影响。类似于表面两层, 第三层O原子和第四层Ni原子也分别发生向内弛豫和向外弛豫, 使d23和d45增大, d34减小。表面重构以后, 整体层间距均小于体相NiO, 除d34外层间距相比于理想表面进一步减小, 其中表面Ni— O键长缩短到0.191 3 nm。三角锥结构键长变短、排列更加紧密, 更能稳定表面的O原子。文献[19]表明, 表面重构使化学配比发生改变, 形成电荷补偿的Octopolar结构, 不管在贫氧还是富氧条件下, 都十分稳定。
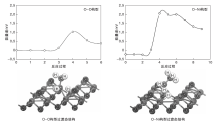
在乙烷活化过程中, 表面O原子发挥至关重要作用。理想表面与重构表面上O原子的H吸附能分别为-5.28 eV和-2.74eV, H吸附能越小, 表明O原子在C— H键断裂过程中越容易与H结合, 越有利于C— H键断裂。理想表面O原子的H吸附能大大低于重构表面, 表明乙烷在这个表面上更容易活化, 与能垒的大小关系相印证。为进一步了解两个表面的不同, 对表面作电子局域函数ELF[27]分析, ELF可用来表征电子的局域化分布特征和成键类型, ELF值为0处电子完全离域, 1.00处电子完全定域; ELF值适中为共价键, 值越大, 离子键成分越多。理想表面与重构表面的ELF图如图2所示。
 | 图2 理想表面与重构表面的ELF图Figure 2 ELF figures of ideal and reconstructed surfaces |
从图2可以看出, NiO固体中Ni— O键不是完全的离子键, 含有一定的共价键成分。理想表面O原子的ELF相对较小, 电子离域化程度较高, Ni— O键的共价键成分更多; 重构表面ELF值较大, 电子更加定域, 离子键成分较多, 表明重构表面O原子与Ni原子的成键更强, 表面更稳定。
乙烷是典型的非极性分子, 在NiO(111)表面为弱物理吸附, 吸附热小, 乙烷分子与表面距离大于0.3 nm, 距离较远, 所以乙烷的吸附构型对其吸附热和C— H键的裂解影响不大。乙烷C— H键断裂后, H和乙基碎片在NiO(111)表面有不同的吸附位点, 不同吸附位点可能会影响C— H键断裂的能垒。表2为H和乙基在NiO(111)表面的吸附位点与反应热和反应能垒的关系。
| 表2 H和乙基在NiO(111)表面的吸附位点与反应热和反应能垒的关系 Table 2 The relationship betweenadsorption sites of radicals, reaction energy and activation barrier on different surfaces of NiO(111) |
由表2可见, 理想NiO(111)表面为O原子, 所以乙烷C— H键裂解后, 碎片吸附到O原子上形成O— H和O— C2H5, 为放热反应, 放出4.29 eV热量。过渡态计算发现, 在这个表面乙烷活化是一个没有能垒的过程, 即这个过程自发进行, 表明理想的NiO(111)表面催化乙烷C— H键断裂活性较好。重构表面同时存在O原子和Ni原子, 乙烷C— H键断裂后H原子碎片可吸附到O上形成羟基(O-H), 或是吸附到Ni的桥位上形成Ni-H-Ni; 研究发现, 乙基碎片只能吸附到Ni上形成Ni-C2H5, 这可能是重构以后表面O原子的性质发生改变, 从而无法与乙基上的C原子成键。在得到的两种构型中, 形成O-H和Ni-C2H5(O-Ni构型), 不管从反应热还是能垒来看, 都要比形成Ni-H和Ni-C2H5(Ni-Ni构型)容易。表明在重构表面, 乙烷C— H键断裂后更倾向于形成O-H和Ni-C2H5, 这步反应需克服1.03 eV能垒, 反应吸热0.55 eV。总体来说, 理想NiO(111)表面活性较好, 可使乙烷自发裂解; 表面重构后催化乙烷C— H键断裂活性降低, 反应需越过1.03 eV能垒。
乙烷在NiO上的活化是一个电子转移过程, 通过对C— H键断裂过程进行Bader charge[28]分析, 能更好了解反应的电子转移情况。图3为H和乙基在NiO(111)表面不同吸附位点反应时的Bader charge图, O-Ni构型表示H原子和乙基的吸附位点分别是O和Ni。
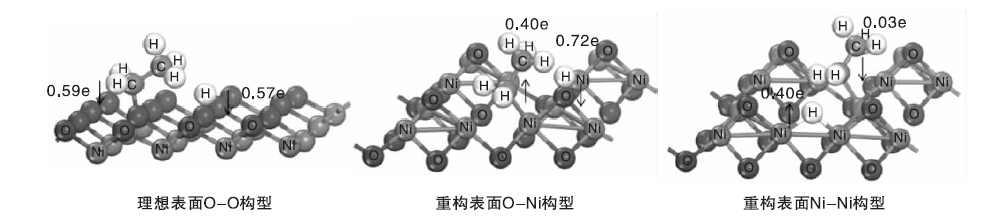 | 图3 H和乙基在NiO(111)表面不同吸附位点反应时的Bader charge图Figure 3 Bader charges of H andethyl radicals reacted on adsorption site of Ni(111) surface |
由图3可见, 在理想表面乙基和H分别向表面转移0.59和0.57个电子; 重构表面上O-Ni构型中, H转移0.72个电子到NiO表面, 乙基从NiO表面得到0.40个电子。在电子得失过程中, 失去电子的是L碱, 得到电子的是L酸, H和乙基分别与表面形成一个类似酸碱对[29]的形式, 这种酸碱相互作用或许能促进烷烃C— H键断裂。在Ni-Ni构型中, 由于H吸附到Ni上, Ni含有较多的3d电子, 所以H从NiO表面得到0.40个电子。
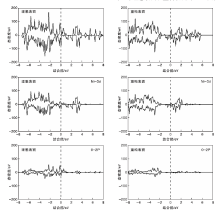
对C— H键断裂过程进行过渡态分析, 发现在过渡态反应过程曲线的能垒点, 不同吸附构型的过渡态结构有明显不同。图4为NiO(111)重构表面不同吸附构型的过渡态反应过程曲线和过渡态结构图。
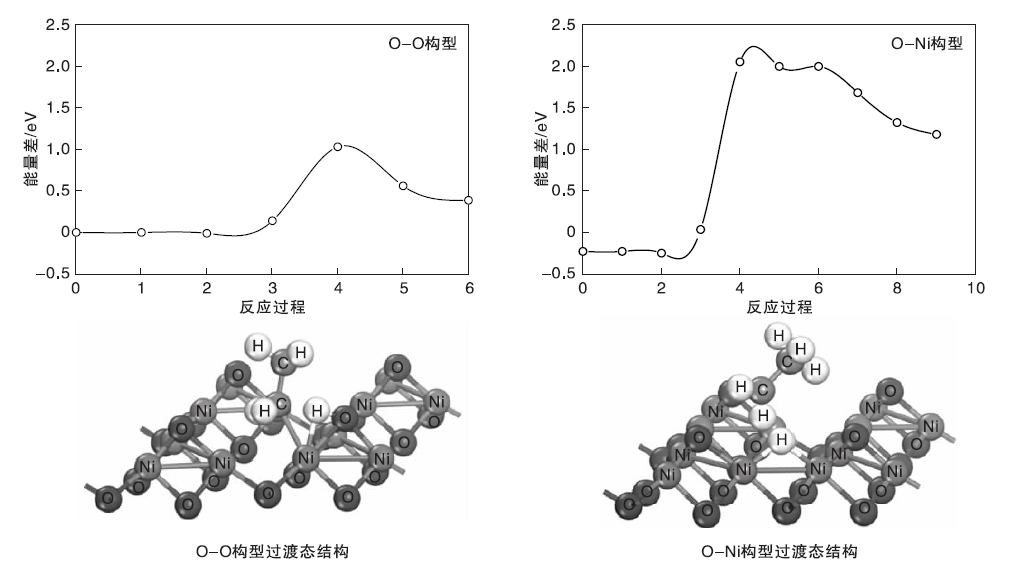 | 图4 NiO(111)重构表面不同吸附构型的过渡态反应过程曲线和过渡态结构图Figure 4 Reaction curves and structures of transition states on NiO(111) reconstructed surface |
由图4可见, 重构表面O-Ni构型的过渡态形成的是一个C2H5-Ni-H-O的4中心结构, 原子之间相互成键, 距离较近, 结构相对比较稳定, 能垒较低。而Ni-Ni构型在过渡态时形成的是Ni-H-Ni和乙基碎片, 乙基碎片与表面距离较远, 不与任何原子成键, 自由度较大, 并且可向不同方向移动, 结构稳定性较差, 能垒相对较高。
通过对理想表面和重构表面进行DOS计算, 可以观察两种表面的电子分布情况。图5为NiO(111)理想表面和重构表面的DOS图。
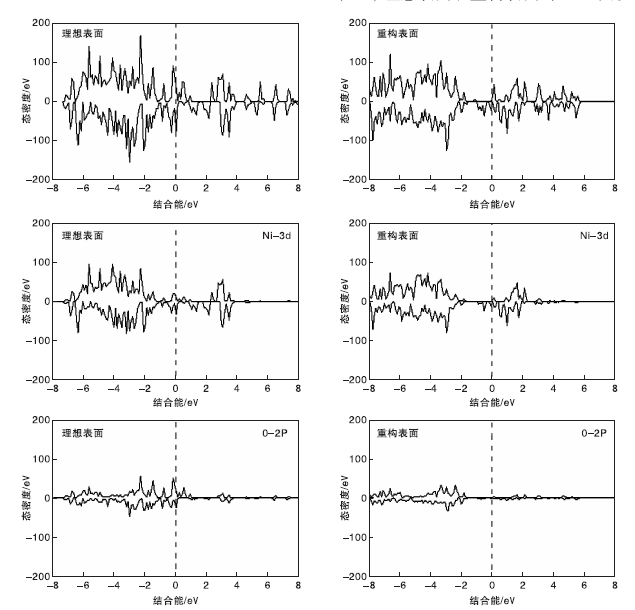 | 图5 NiO(111)不同表面的DOS图Figure 5 DOS of NiO(111) different surfaces |
由图5可见, 两种表面的电子均越过费米能级, 表面没有带隙, 呈金属性质; 费米能级左侧价带(VB)电子主要是Ni-3d和O-2p轨道贡献, 费米能级右侧导带(CB) 中O-2p电子非常少, 主要是Ni-3d轨道的电子。因为乙烷活化是在表层原子上进行, 所以表层O原子的2p轨道DOS图能更直观地表明不同O原子的性质。
图6为NiO(111)表面O-2p轨道DOS图。
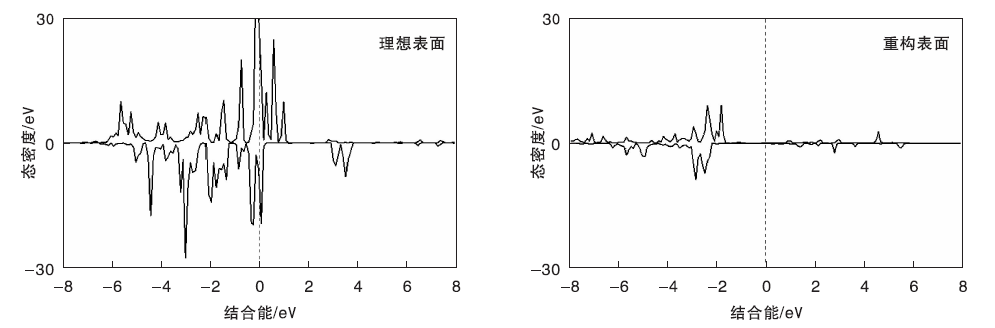 | 图6 NiO(111)表面O-2p轨道DOS图Figure 6 DOS of O-2p orbital on NiO(111) surfaces |
由图6可见, 两种表面最外层O原子的O-2p轨道电子存在明显的区别, 理想表面中O-2p电子主要分布在价带和导带底, 电子越过费米能级, 性质较为活泼; 而重构表面中O-2p电子主要分布在价带底部和中部, 费米能级附近几乎无电子, 不利于反应中进行电子转移, 重构表面乙烷较难活化。
(1) 乙烷在理想NiO(111)表面上的C— H键裂解是一个没有能垒的自发过程; 表面重构使表面形成三角锥结构, 并且表面向内收缩, 层间距减小, 表面更加稳定。
(2) 重构表面上乙烷C— H键活化较为困难, 能垒较高。通过对反应进行结构和电子分析发现, 理想表面的O原子更容易吸附H原子, O-2p轨道电子越过费米能级, 性质较为活泼, 有利于C— H键的断裂。
(3) 重构表面O原子的H吸附能较大, 电子更加局域化, 费米能级附近几乎无O-2p轨道电子分布, 乙烷活化较为困难。
(4) 在重构表面上, 乙烷C— H键断裂可形成两种不同构型, 其中形成O-H和Ni-C2H5的能垒更小, 这可能与过渡态时形成C2H5-Ni-H-O的四中心结构和电子转移时形成的酸碱对有关。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|