{kind=link}
{kind=link}
{kind=link}
甲醛在Pd(111)与Pd(100)晶面上氧化的计算化学研究
[任宇1 , 宋卫余1 , 刘坚1 , 赵震1, 2, *  ]
]
]
|
|


作者简介:任宇,1992年生,男,在读博士研究生,主要从事贵金属催化甲醛氧化以及电催化还原二氧化碳方面的计算化学研究。
使用密度泛函理论对甲醛分子在Pd(111)晶面与Pd(100)晶面的氧化反应机理进行研究,结果表明,Pd(100)晶面相比于Pd(111)晶面更有利于甲醛分子的氧化,为进一步合理设计甲醛氧化贵金属催化剂提供了理论依据。
Reaction mechanism for formaldehyde oxidation on Pd(111) and Pd(100) facets is studied by density functional theory.Results show that Pd(100) crystal surface is more conducive to oxidation of formaldehyde molecules than Pd(111) crystal surface.It provides theoretical basis for further rational design of precious metal catalysts for the title reaction.
甲醛是一种常见的化学物质, 常见于家庭和办公室, 室内空气中甲醛已成为影响人类身体健康的主要污染物[1]。2017年10月27日, 世界卫生组织国际癌症研究机构公布的致癌物清单初步整理参考, 甲醛在一类致癌物清单中。常温下脱除甲醛且不产生二次污染受到了广泛研究。其中贵金属催化剂由于良好的低温活性具有极大的应用前景, Zhang C等[2]采用Pt、Rh、Pd和Au等贵金属催化甲醛氧化取得良好的活性, 证明Pt/TiO2催化剂在室温可以将甲醛完全转化。Huang H等[3]报道了二氧化钛载体负载贵金属钯催化剂在室温下将甲醛完全转化为水和二氧化碳, 催化剂活性与金属表面的性质密切相关。
本文采用密度泛函理论计算(DFT)方法探究甲醛在Pd(111)晶面及Pd(100)晶面的氧化机理, 揭示金属钯催化甲醛氧化的构效关系。
使用Vasp软件包[4, 5]计算, 采用周期性赝势平面波的自旋极化密度泛函计算方法, 通过GGA-PBE[6]方法描述电子相互关联效应, 采用PAW赝势描述原子核与价电子间的相互作用[7], 同时考虑自旋极化效应, 其中截断能为400 eV, 采用CI-NEB[8, 9]方法计算过渡态, 收敛标准设置为0.05 eV, 真空层厚度采用1.2 nm, 确保上下两层不会产生相互作用力。Pd(111)与Pd(100)的模型采用4× 4大小4层厚的模型, 当优化结构时固定下面两层弛豫上面两层。布里渊区K点选择3× 3× 1。通过CI-NEB方法求每一基元步骤的过渡态, 并验证过渡态有且仅有一个虚频。
HCHO、O2、H2O和CO在Pd(111)和Pd(100)上的吸附能通过方程(1)得出:
Eads=Ea+b-(Ea+Eb)(1)
式中, Eads为吸附能, eV; Ea+b为表面吸附分子后的能量, eV; Ea为表面能, eV; Eb为吸附分子的能量, eV。
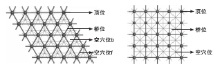
图1为Pd(111)晶面与Pd(100)晶面上的吸附位。
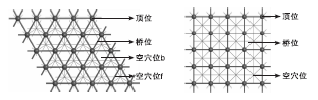 | 图1 Pd(111)晶面与Pd(100)晶面上的吸附位Figure 1 Adsorption site on Pd(111) and Pd(100) surface |
由图1可见, 当小分子吸附在贵金属Pd晶面上时, Pd(100)晶面有顶位、桥位以及空穴位三种吸附位置, 而Pd(111)晶面有顶位、桥位、空穴位b和空穴位f四种吸附位置。通过计算分子在不同位置的吸附能确定最稳定构型, 即采用吸附能最大的构型。
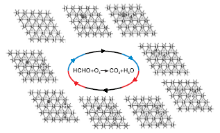
图2为甲醛分子在Pd(111)晶面上的氧化过程。
由图2可见, 当甲醛分子吸附在Pd(111)表面时, 甲醛分子可以稳定吸附在桥位或空穴位b, 吸附能分别为1.1 eV和1.14 eV, 表明Pd(111)晶面对甲醛分子有较强的吸附能力, 之后甲醛分子分别经过0.37 eV和0.42 eV能垒断裂一个氢原子到旁边的空穴位b, 此时断裂一个氢原子所形成的构型相同, 然后CHO需要0.2 eV能垒脱掉第二个氢原子形成一氧化碳分子, 可以看出, Pd(111)表面有利于甲醛活化脱氢过程。氧气分子吸附在桥位上, 吸附能1.42 eV, 表明氧气容易吸附在Pd(111)表面, 计算得出氧气分子不需要能垒就可以直接裂解成两个氧原子吸附在空穴位b上, 证明Pd(111)晶面对氧气的裂解起促进作用, 氢原子在金属Pd表面从一个空穴位迁移到另一个空穴位也无需能垒。其中一个氧原子与一氧化碳分子需要1.01 eV能垒形成二氧化碳分子, 另一个氧原子先后与两个氢原子反应生成水分子, 所需能垒分别为0.78 eV和0.52 eV。最后二氧化碳和水分子从表面脱附掉完成整个循环。从整个过程可以看出, 甲醛脱氢过程及氧气裂解过程在Pd(111)晶面容易进行, 而一氧化碳分子与氧原子形成二氧化碳分子的过程为整个反应的决速步骤。Jing M等[10]研究甲醛氧化时也得到类似结果。
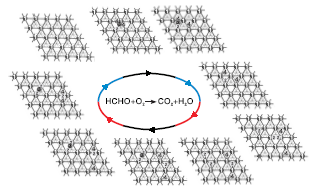 | 图2 甲醛分子在Pd(111)晶面上的氧化过程 1.Pd原子; 2.氧原子; 3.碳原子; 4.氢原子Figure 2 Oxidation steps of formaldehyde on Pd(111) surface |
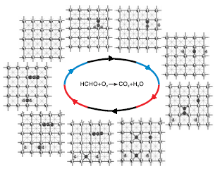
图3为甲醛分子在Pd(100)晶面上的氧化过程。
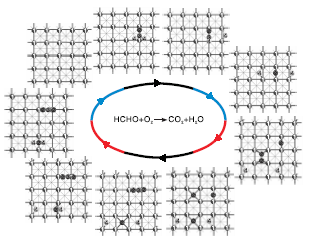 | 图3 甲醛在Pd(100)晶面上的氧化过程 1.Pd原子, 2.氧原子, 3.碳原子, 4.氢原子Figure 3 Oxidation steps of formaldehyde on Pd(100) surface |
由图3可见, 甲醛分子吸附在Pd(100)晶面上桥位及空穴位时吸附能均为0.67 eV, 比Pd(111)晶面吸附甲醛分子的能力弱, 之后甲醛分子先后经过0.19 ev和0.2 eV能垒脱掉两个氢原子生成一氧化碳分子, 表明Pd(100)晶面也能非常容易地活化甲醛脱氢, 并且比Pd(111)晶面活化能力更高, 然后氧气以1.5 eV吸附能吸附在Pd(100)表面的桥位上, 同样也不需要能垒裂解为两个氧原子吸附到空穴位上, 一个氧原子与一氧化碳分子经过0.82 eV能垒生成二氧化碳分子, 另一个氧原子先后与两个氢原子反应生成水, 生成羟基过程的能垒为0.31 eV, 羟基生成水的过程能垒为0.5 eV。一氧化碳生成二氧化碳过程为整个反应决速步骤, Pd(100)晶面也有利于甲醛分子活化脱氢和氧气裂解过程, 通过与Pd(111)晶面上的过程对比可知, 各个基元步骤所需的能垒低于甲醛分子在Pd(111)晶面上, 可以看出, Pd(100)晶面比Pd(111)晶面有更好的催化甲醛氧化活性。可能与Pd(100)晶面原子配位数比Pd(111)晶面原子配位数低有关。各个基元步骤的过渡态及能垒见表1, 能量单位为eV。
| 表1 甲醛氧化每个基元步骤的过渡态及能垒 Table 1 Transition states and energy barriers for each elementary step of formaldehyde oxidation |
| 续表1 Xu Table |
(1) 甲醛分子与氧气分子吸附在金属Pd(111)晶面时更容易吸附在空穴位, 由于次表面原子的影响, 吸附在空穴位b比空穴位f更稳定, 并且Pd(111)晶面吸附甲醛分子的能力强于Pd(100)晶面。
(2) 甲醛分子在Pd(111)晶面与Pd(100)晶面氧化过程的速率控制步骤均为一氧化碳分子与氧原子生成二氧化碳分子的过程, 并且Pd(111)晶面与Pd(100)晶面有利于甲醛分子的脱氢活化过程以及氧气裂解过程。
(3) 甲醛分子在Pd(100)晶面上氧化时各个基元步骤的能垒比在Pd(111)晶面低, 从动力学角度考虑, 相对于Pd(111)晶面, 甲醛分子更易于在Pd(100)晶面上完全氧化。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|