

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
胺功能化MIL-101(Cr)@SiO2@Fe3O4催化剂及其Knoevenagel反应性能
[蒋赛1, 2 , 代武军1 , 汤之强1 , 李金兵1 , 季生福2,*  ]
]
]
|
|


作者简介:蒋赛,1988年生,男,山东省聊城市人,博士,工程师,研究方向为工业催化。
采用溶剂热法在纳米SiO2@Fe3O4磁性颗粒表面原位合成MIL-101(Cr),制备磁性MIL-101(Cr)@SiO2@Fe3O4催化剂。采用甲胺、乙二胺和丁二胺对制备的磁性催化剂进行功能化,得到胺功能化NH2-MIL-101(Cr)@SiO2@Fe3O4催化剂。利用XRD、FT-IR、BET、SEM、TEM和VSM等对催化剂结构进行表征,评价胺功能化NH2-MIL-101(Cr)@SiO2@Fe3O4催化剂对糠醛和氰乙酸乙酯Knoevenagel缩合反应性能和重复使用性能,考察反应条件与催化性能的关系。结果表明,制备的新型胺功能化NH2-MIL-101(Cr)@SiO2@Fe3O4催化剂具有MIL-101(Cr)的结构特征和良好的超顺磁性能,对糠醛和氰乙酸乙酯Knoevenagel缩合反应表现出很好的催化性能,其中,乙二胺功能化30%MIL-101(Cr)@SiO2@Fe3O4催化剂对Knoevenagel 缩合反应的性能最佳,在反应温度40 ℃和反应时间1 h条件下,氰乙酸乙酯转化率为97.0%,产物选择性接近100%。反应后磁性催化剂可以通过外磁场容易进行分离,重复使用5次,氰乙酸乙酯转化率仍大于93%。
The magnetically MIL-101(Cr)@SiO2@Fe3O4 catalysts with different MIL-101(Cr) contents were synthesized using in situ method of encapsulating MIL-101(Cr) on magnetic SiO2@Fe3O4 nanoparticles surface.Then methylamine,ethylenediamine and butanediamine were used as functional groups for MIL-101(Cr)@SiO2@Fe3O4 and novel NH2-MIL-101(Cr)@SiO2@Fe3O4 catalysts were prepared.Structure of the catalysts was characterized by XRD,FT-IR,BET,SEM,TEM and VSM techniques.Catalytic performance and recovery properties of the catalysts for the Knoevenagel reaction of furfural with ethyl cyanoacetate were evaluated.The relationship of reaction conditions and catalytic performance were also investigated.The results showed that all three amines functionalized catalysts had the structure of MIL-101(Cr) and good superparamagnetism.The magnetic NH2-MIL-101(Cr)@SiO2@Fe3O4 catalysts exhibited well catalytic performance for the Knoevenagel reaction of furfural with ethyl cyanoacetate.Among them, the 30%MIL-101(Cr)@SiO2@Fe3O4 catalyst was the best one.Conversion of ethyl cyanoacetate could reach 97.0% and selectivity of product could reach~100% at reaction temperature of 40 ℃ and 1 h reaction time.After reaction,the catalysts could be easily separated from the reaction mixture by an external magnet.The recovery catalyst could be reused for five times, and conversion of ethyl cyanoacetate kept over 93%.
Knoevenagel反应是一类羰基化合物与活性亚甲基化合物的脱水缩合反应, 常用于C 
金属有机骨架化合物(MOFs)是一种由有机配体和金属离子连接而成的介孔材料。由于具有巨大的孔隙度和孔的几何形状、中心金属、不饱和配位点及可负载活性组分, 使MOFs在催化领域具有很大的应用价值和潜力[8, 9]。在丙二腈和苯甲醛Knoevenagel缩合反应中, Valvekens P等[10]以Mg、Co、Ni、Cu和Zn等为金属中心, 2, 5-dioxidoterephthalate为配体, 制备了M2dobdc催化剂并应用到Knoevenagel缩合反应中, 但催化效果不佳, 性能最好的Ni2dobdc催化剂反应2 h, 收率仅69%。Luo Qunxing等[11]用Cu3(BTC)2 负载碱性离子液体ABIL-OH为催化剂进行反应, 产物选择性100%, 反应3 h, 苯甲醛转化率可达100%, 但该催化剂重复使用性能较差, 重复使用6次, 转化率降至80%。Gascon J等[12]首先提出具有不饱和氨基基团的MOF材料如IRMOF-3和Amino-MIL-53(Al)在Knoevenagel缩合反应中表现出与固体碱催化剂一样稳定的催化性能, 并从碱性强弱角度解释反应可能进行的两种机理。Cortese R等[13]在此基础上进行了IRMOF-3催化Knoevenagel缩合反应的DFT研究, 进一步证实了-NH2 为活性中心及该催化剂具体的催化机理。此后大量氨基功能化MOF材料如NH2-MIL-101(Al)[14, 15]、NH2-MIL-101(Fe)[16]和UiO-66-N
将磁性与催化剂相结合是催化剂制备研究的重要手段[19]。具有超顺磁性的催化剂在固-液相反应中, 附加外部磁场时具备快速分离的优势, 大大简化了操作[20]。Ji Junhong等[21]成功制备磁性Cu/Fe3O4@SiO2催化剂, 可在室温下将低浓度甲醛催化转换为氢气, 催化剂重复使用8次, 催化剂催化性能未见明显下降。Liu Hongfei等[22]制备了具有核壳结构的TiO2/SiO2@Fe3O4光催化剂, 并应用于甲基橙和亚甲基蓝染料降解中, 取得了较好效果。Li Qingyuan等[23]成功制备了磁性Cu-BTC@SiO2@Fe3O4催化剂并应用于Pechmann反应中, 1-萘酚转化率96%, 目标产物选择性98%, 催化剂重复使用5次后, 1-萘酚转化率保持在90%以上。Jiang Sai等[24]制备了磁性MIL-53(Al)@SiO2@Fe3O4催化剂, 在苯甲酰氯和2-甲基吲哚的F-C酰基化反应中取得良好结果。
本研究尝试将MOFs材料与具有超顺磁性纳米材料Fe3O4相结合, 在磁核表面原位生长出MIL-101(Cr), 制备具有核壳结构的MIL-101(Cr)@SiO2@Fe3O4复合材料。采用后合成方法, 用不同碳链长度的胺类功能化, 制备不同胺类嫁接MOF磁性复合NH2-MIL-101(Cr)@SiO2@Fe3O4材料。采用XRD、FT-IR、TEM、SEM和VSM等表征催化剂结构。研究催化剂在氰乙酸乙酯和糠醛Knoevenagel缩合反应中的催化性能, 考察不同胺类物质功能化对催化活性的影响以及反应温度和催化剂用量等对反应的影响。
FeCl3· 6H2O、醋酸钠、氨水, 分析纯, 国药集团化学试剂有限公司; 聚乙烯吡咯烷酮(K23-2)、正硅酸乙酯(98%)、H2BDC(99%)、硅酸乙酯(98%), 阿拉丁试剂; Cr(NO3)3· 9H2O(99.0%)、甲胺、乙二胺、丁二胺, 分析纯, 阿拉丁试剂。
(1) 磁性SiO2@Fe3O4合成。参照文献
(2) 不同MOF含量MIL-101(Cr)@SiO2@Fe3O4制备。参照文献[26]水热合成法制备无氟MIL-101(Cr)。将0.4 g的Cr(NO3)3· 9H2O (1 mmol)溶解于4.8 mL去离子水中, 加入0.16 g的H2BDC(1 mmol), 得到均匀MOF混合物。分别将0.5 mmol、1 mmol、1.5 mmol和2 mmol的SiO2@Fe3O4纳米磁核加入上述MOF混合物体系中, 超声15 min以将所有组分尽可能分散均匀。将反应物放入Teflon内衬的自压力反应釜中, 恒温220 ℃反应8 h, 室温冷却。在外加磁场下快速分离得到固体, 用丙酮和无水乙醇加热冷凝回流12 h, 进行纯化, 磁性分离, 将得到的固体粉末真空60 ℃干燥过夜, 制得20%MIL-101(Cr)@SiO2@Fe3O4、30%MIL-101(Cr)@SiO2@Fe3O4、40%MIL-101(Cr)@SiO2@Fe3O4和50%MIL-101(Cr)@SiO2@Fe3O4的磁性复合材料。
(3) 不同胺类功能化NH2-MIL-101(Cr)@SiO2@Fe3O4催化剂制备。综合文献
XRD表征采用日本理学公司D/MAX 2500 VB 2+/PC型X射线衍射仪, 扫描范围5° ~80° 。
SEM采用德国蔡司公司SUPRA55电子扫描显微镜。
TEM采用日本电子仪器公司JEM-2100高分辨的透射电子显微镜。
催化剂的孔径分布和比表面积测试采用美国麦克仪器公司SAP2020M。测试前样品先在100 ℃脱气12 h, BET 法计算样品的比表面积, BJH法计算样品的孔径和孔体积。
元素分析采用德国Elemeraor公司Vario EL cube分析仪。
FT-IR表征采用德国布鲁克公司Tensor-27型傅立叶红外光谱仪, 波数(4 000~600) cm-1。
催化剂的饱和磁化强度曲线通过Lake Shore振动样品磁强计(VSM) 7410测量获得。
氰乙酸乙酯和糠醛Knoevenagel缩合反应在带有冷凝回流和搅拌装置的100 mL三口烧瓶中进行。瓶内依次加入5 mL甲苯作为溶剂、7 mmol氰乙酸乙酯和糠醛为反应物、0.5 mL十二烷作为内标物及一定量催化剂。反应在40 ℃搅拌条件下进行, 反应结束后自然冷却, 利用磁性分离, 得到的固体催化剂用乙醇冲洗, 120 ℃真空干燥12 h后称重, 计算催化剂回收率。按上述反应步骤进行催化剂的重复使用性能评价。磁性分离得到的反应液中加入无水硫酸钠除去反应生成的水, 然后定量在北京东西分析仪器公司GC-4000A型气相色谱仪上进行分析, 色谱柱为Factor Four:Capillary Column VF-1ms, 60 m× 0.25 mm× 0.25 μ m, 采用内标法定量, 计算氰乙酸乙酯转化率和产物选择性。
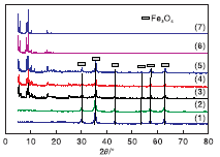
对合成的MIL-101(Cr)、NMM-1、NMM-2和NMM-3进行XRD表征, 结果见图1。
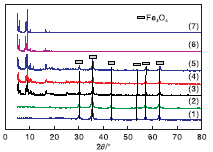 | 图1 模拟及合成的不同样品的XRD图 (1)Fe3O4; (2)SiO2@Fe3O4; (3)NMM-1; (4)NMM-2; (5)NMM-3; (6)合成的 MIL-101(Cr); (7)模拟MIL-101(Cr) |
从图1可以看出, 纳米Fe3O4表面包覆SiO2后, XRD图没有明显变化。模拟标准MIL-101(Cr)与合成的MIL-101(Cr)的特征峰对应程度较好。NMM-1、NMM-2和NMM-3的XRD图中, 在30.0° 、35.4° 、43.0° 、53.4° 、56.9° 和62.5° 出现的衍射峰归属为典型的立方体尖晶石结构Fe3
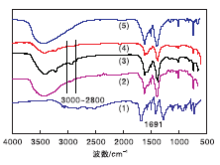
图2给出了样品的FT-IR图谱。从图2可以看出, H2BDC谱图上可以看到1 691 cm-1处有强振动峰, 归属为典型的H2BDC中-COOH伸缩振动峰。而合成的MIL-101(Cr)、NMM-1、NMM-2和NMM-3在约1 691 cm-1处没有吸收峰, 表明合成的MOF样品中不存在自由H2BDC分子, 即-COO基团与金属Cr结合, NMM-1、NMM-2和NMM-3催化剂中1 617 cm-1和1 395 cm-1处的峰归于对苯二甲酸中羧酸根的羰基(-COO-)不对称伸缩振动和对称伸缩振动峰, 1 020 cm-1和753 cm-1处归属于芳香环上的γ (C-H)和δ (C-H), 650 cm-1处归于Cr-O振动峰, 与MIL-101(Cr)的谱图相对应。因此, 可以确定NMM-1、NMM-2和NMM-3催化剂均含有MIL-101(Cr)结构。
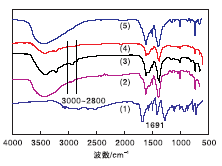 | 图2 样品的FT-IR谱图Figure 2 FT-IR spectra of samples (1)H2BDC; (2)NMM-1; (3)NMM-2; (4)NMM-3; (5)合成的MIL-101(Cr) |
从图2(3)和图2(4)谱图可以明显看到属于胺类物质的N-H伸缩振动峰[(3 300~3 150) cm-1]和脂肪族C-H的伸缩振动峰[(3 000~2 800) cm-1][27], 虽然NMM-1催化剂样品[图2(2)]没有这么明显, 但均表明胺类物质成功嫁接。与合成的MIL-101(Cr)谱图对比, 3个催化剂上的脂肪族C-H伸缩振动峰峰值增加, 只有当分子与L酸酸性中心配位时才可被观察到, 说明3种胺类物质嫁接在chromium(Ⅲ )不饱和活性位点上
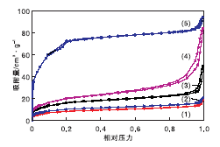
对NMM-1、NMM-2、NMM-3、30%MIL-101(Cr)@SiO2@Fe3O4和合成的MIL-101(Cr)进行N2吸附-脱附表征, 所有样品均在77 K下测试, 结果见图3。
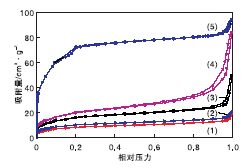 | 图3 样品的N2吸附-脱附谱图Figure 3 N2 adsorption-desorption spectra of samples (1)NMM-3; (2) NMM-2; (3) NMM-1; |
(4) 30%MIL-101(Cr)@SiO2@Fe3O4; (5) 合成的MIL-101(Cr)
由图3可见, 样品曲线属于吸附等温线分类中的类型Ⅱ , 即反S型曲线, 在低压区发生的是Ⅰ 型吸附, 在后半段由于发生多分子层吸附或毛细凝聚会呈现反S, 体现出中孔和微孔材料的特征。
表1为样品的孔结构参数。从表1可以看出, 纯MIL-101(Cr)的比表面积为3 086.1 m2· g-1, 基本符合文献[16]报道。而30%MIL-101(Cr)@SiO2@Fe3O4复合材料可以认为磁核和MOF共同贡献了比表面积, 略大于纯MOF材料的三分之一。而NMM-1、NMM-2和NMM-3催化剂的比表面积、孔径和孔体积均有不同程度下降, 分析原因为, 功能化过程本身涉及到加热, 对材料骨架有一定程度的影响; 胺类分子占据了MOF结构中的不饱和活性位点, 会造成孔径和孔体积的减小
| 表1 样品的孔结构参数 Table 1 Pore characteristics of samples |
图4和图5为NMM-1、NMM-2和NMM-3催化剂的SEM和TEM照片。从图4和图5可以看出, 3种催化剂形貌颗粒大小比较均匀, 均是直径约为150 nm的微球, 均具备规则的核壳结构[24], 外层包覆的MOF壳层厚度约为(5~10) nm, 深色的复合磁性载体SiO2@Fe3O4被包覆在浅色MIL-101(Cr)的里面。
 | 图4 NMM-1、NMM-2和NMM-3催化剂的SEM照片Figure 4 SEM images of NMM-1, NMM-2 and NMM-3 catalysts |
 | 图5 NMM-1、NMM-2和NMM-3催化剂的TEM照片Figure 5 TEM images of NMM-1, NMM-2 and NMM-3 catalysts |
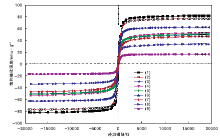
图6为样品的VSM图。从图6可以看出, 所有样品均呈现超顺磁性特性, 并且饱和磁化强度随着催化剂中MIL-101(Cr)含量的增加而减小。其中, Fe3O4的饱和磁化强度为82.0 emu· g-1, SiO2@Fe3O4的饱和磁化强度略降, 为76.9 emu· g-1, 这主要是包覆了薄层SiO2所致。
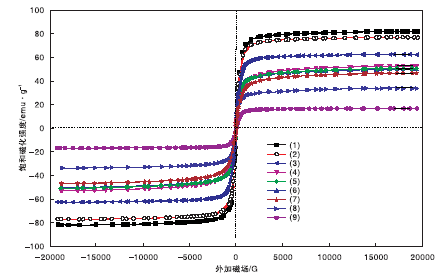 | 图6 样品的VSM图Figure 6 VSM profiles of all samples (1)Fe3O4; (2)SiO2@Fe3O4; (3)20%MIL-101(Cr)@SiO2@Fe3O4; (4)30%MIL-101(Cr) @SiO2@Fe3O4; (5)NMM-3; (6)NMM-2; (7)NMM-1; (8)40%MIL-101(Cr) @SiO2@Fe3O4; (9)50%MIL-101(Cr)@SiO2@Fe3O4 |
合成的4种不同MOF含量(20%~50%)的磁性材料按照包覆量从小到大, 其饱和磁化强度依次为62.5 emu· g-1、52.6 emu· g-1、33.7 emu· g-1和16.7 emu· g-1, 表明随着MOF包覆量的增加, 材料的磁化强度呈负相关
利用糠醛和氰乙酸乙酯的Knoevenagel缩合反应对催化剂进行评价, 考察不同催化剂在不同条件下的催化活性。
反应体系中未添加催化剂时, 氰乙酸乙酯转化率低于2%, 考虑到误差因素, 可以认为在没有催化剂条件下, 不发生反应。反应中目标产物选择性为100%, 与大量关于MOF催化Knoevenagel缩合反应的研究结果吻合, 体现了MOF作为催化剂时选择性高的突出优势[11, 12]。Opanasenko M等
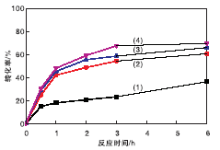
在催化剂用量0.2 g(1.17 mmol)、氰乙酸乙酯7 mmol、苯甲醛7 mmol、溶剂甲苯5 mL、正十二烷0.5 mL和反应温度40 ℃条件下, 考察未经胺化不同MOF含量催化剂上不同反应时间对转化率的影响, 结果见图7。从图7可以看出, 未经胺嫁接修饰的MIL-101(Cr)@SiO2@Fe3O4催化剂对糠醛和氰乙酸乙酯的Knoevenagel缩合反应也有一定催化活性。其中, 50%MIL-101(Cr)@SiO2@Fe3O4催化剂活性最高, 反应6 h, 氰乙酸乙酯转化率接近70%, 与Hartmann M等[35]的报道相呼应。在保证催化活性前提下, 要求催化剂具有良好的超顺磁性。而磁性强度与催化剂中MOF比例负相关。综合考虑, 选择MOF质量分数30%的催化剂, 其催化活性略逊于MOF质量分数40%和50%, 但远高于MOF质量分数20%, 其顺磁性远好于MOF质量分数40%和50%。选择用3种胺类进行嫁接后修饰30%MIL-101(Cr) @SiO2@Fe3O4材料。
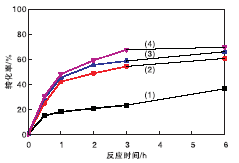 | 图7 不同MOF含量催化剂上反应时间对转化率的影响Figure 7 Effects of MOF contents on conversion over different catalysts (1)20%MIL-101(Cr)@SiO2@Fe3O4; (2)30%MIL-101(Cr)@SiO2@Fe3O4; (3)40%MIL-101(Cr)@SiO2@Fe3O4; (4)50%MIL-101(Cr)@SiO2@Fe3O4 |
在催化剂用量0.2 g(1.17 mmol)、氰乙酸乙酯7 mmol, 、苯甲醛7 mmol, 、溶剂5 mL甲苯、正十二烷0.5 mL和反应温度40 ℃条件下, 考察3种胺功能化NH2-30%MIL-101(Cr)@SiO2@Fe3O4催化剂催化性能, 结果见图8。
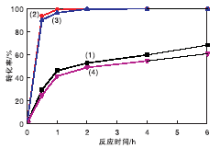
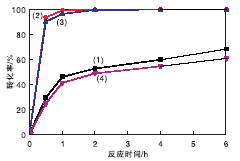 | 图8 不同胺功能化催化剂上反应时间对转化率的影响Figure 8 Effect of reaction time on conversion over different catalysts (1)NMM-1; (2)NMM-2; (3)NMM-3; (4)30%MIL-101(Cr)@SiO2@Fe3O4 |
由图8可见, 对于糠醛和氰乙酸乙酯Knoevenagel缩合反应, 催化剂的催化活性顺序为:NMM-2> NMM-3> NMM-1> 30%MIL-101(Cr)@SiO2@Fe3O4。表明反应中催化性能的优劣由催化剂中-NH2中心数量决定
2.7.1 反应温度
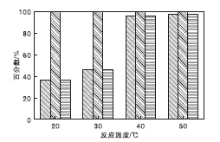
在NMM-2催化剂用量0.2 g(1.17 mmol)、氰乙酸乙酯7 mmol、苯甲醛7 mmol、反应时间2 h、溶剂甲苯5 mL和正十二烷0.5 mL条件下, 考察不同反应温度下NMM-2催化剂的催化性能, 结果见图9。
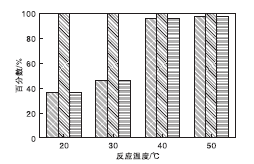 | 图9 不同反应温度下NMM-2催化剂的催化性能Figure 9 Catalytic property at different temperature over NMM-2 catalyst |
由图9可知, Knoevenagel缩合反应对温度的变化非常敏感, 反应条件非常温和, 室温下即可反应。相同反应条件下, 随着反应温度的升高, 转化率呈上升趋势。反应温度40 ℃和50 ℃, 氰乙酸乙酯转化率较高, 分别为95.8%和97.4%, 与Burgoyne A R等[36]的结果吻合。不同反应温度条件下, 产物经色谱检测, 均未出现副产物, 选择性100%。因此, 适宜的反应温度为40 ℃。
2.7.2 催化剂用量
在氰乙酸乙酯7 mmol、苯甲醛7 mmol、溶剂甲苯5 mL、正十二烷0.5 mL、反应温度40 ℃和反应时间2 h条件下, 考察不同NMM-2催化剂用量对反应性能的影响, 结果见表2。
| 表2 不同NMM-2催化剂用量对反应性能的影响 Table 2 Effect of NMM-2 catalyst dosage on catalytic performance |
从表2可以看出, 随着催化剂用量的增加, 氰乙酸乙酯转化率呈先增加后趋于稳定的趋势, 与Li Qingyuan等[37]在使用复合磁性ZIF-8@SiO2@Fe3O4催化剂研究Knoevenagel缩合反应得到的规律吻合, 即催化活性完全取决于复合磁性材料中MOFs含量。催化剂用量0.2 g(1.17 mmol)时, 氰乙酸乙酯转化率99.2%, 继续增加催化剂用量, 氰乙酸乙酯转化率变化不大。可见催化剂用量增加时, 催化剂活性中心增加, 此时反应活性有所提高。但催化剂用量增加到一定程度时, 再增加催化剂用量, 反应中心已经饱和, 反应速率提高不明显。因此, 选择最佳催化剂用量为0.2 g(1.17 mmol)。
2.7.3 溶 剂
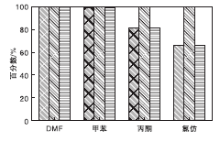
选取一些常见的不同极性有机溶剂作为考察对象, 分别采用5 mL的氯仿、丙酮、甲苯和DMF作为溶剂进行评价。在NMM-2催化剂用量0.2 g(1.17 mmol)、氰乙酸乙酯7 mmol、苯甲醛7 mmol、反应时间2 h和反应温度40 ℃条件下, 考察不同溶剂对反应性能的影响, 结果见图10。从图10可以看出, DMF作溶剂时, 活性最强, 反应2 h, 氰乙酸乙酯转化率接近100%。可以认为, 活泼性亚甲基化合物氰乙酸乙酯由于强极性溶剂的离子化而产生负碳离子, 此时, 带有碳负离子的氰乙酸乙酯则可进攻苯甲醛, 容易发生亲核反应, 即DMF溶剂可以与活泼氢形成氢健.然后生成负碳离子与苯甲醛发生亲核反应
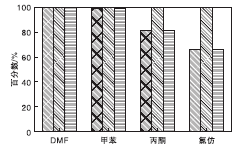 | 图10 不同溶剂对反应性能的影响Figure 10 Effect of solvents on catalytic activity |
2.7.4 反应底物
在NMM-2催化剂用量0.2 g(1.17 mmol)、底物1用量7 mmol、底物2用量7 mmol、甲苯5 mL和正十二烷0.5 mL条件下, 考察反应底物对反应性能的影响, 结果见表3。由表3可以看出, 底物为苯甲醛时, 反应1 h, 氰乙酸乙酯转化率89.1%(序号1)。底物为对氯苯甲醛时, 氰乙酸乙酯转化率95.3%(序号3), 表明苯环上带有吸电子基能促使缩合反应的进行, 有利于形成碳负离子的氰乙酸乙酯亲核进攻苯甲醛进行亲核加成, 与Neogi S等
| 表3 不同反应底物对反应性能的影响 Table 3 Effect of reactants on catalytic activity |
2.7.5 催化剂重复使用性能
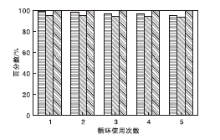
在NMM-2催化剂用量0.2 g(1.17 mmol)、甲苯5 mL、反应时间2 h和反应温度40 ℃条件下, 考察催化剂重复使用性能, 结果见图11。
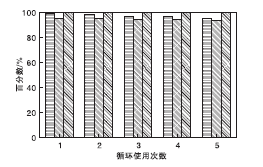 | 图11 催化剂重复使用性能Figure 11 Catalyst recycling studies |
从图11可以看出, 催化剂重复使用5次, 催化剂回收率分别为99.3%、98.1%、97.0%、96.6%和95.2%; 氰乙酸乙酯转化率分别为95.0%、94.8%、94.6%、94.1%和93.8%, 选择性保持100%。在催化剂回收方面, 相较于Xue Bing等[4]报道的传统固体酸性催化剂或Nguyen L T L等
(1) 通过原位合成和超声辅助的方法制备了不同MOF含量(20%~50%)的MIL-101(Cr)@SiO2@Fe3O4复合磁性材料。采用后合成功能化方法, 在复合磁性材料中MOF部分骨架结构中金属不饱和活性位点上成功嫁接不同胺类物质作为催化活性中心。
(2) XRD、FT-IR、BET、SEM、TEM和VSM等表征表明, 甲胺、乙二胺和丁二胺功能化的3种磁性催化剂NMM-1、NMM-2和NMM-3在保持超强顺磁性前提下, 仍具有良好的MOF晶体结构, 催化剂整体呈现出明显的核-壳结构, 催化活性优于未经功能化的MIL-101(Cr)@SiO2@Fe3O4复合材料, 表明胺类物质提供的碱性中心有利于反应性能的提高。
(3) 采用乙二胺功能化NMM-2为催化剂时, 以呋喃甲醛和氰乙酸乙酯为模型反应对催化剂进行评价。反应在比较温和条件下即可进行, 呋喃甲醛与氰乙酸乙酯物质的量比为1∶ 1, 反应温度40 ℃和反应时间1 h条件下, 氰乙酸乙酯转化率为97.0%, 选择性接近100%。NMM-2催化剂优于常见的固体酸和分子筛等非均相催化剂, 并具备优异的超顺磁性, 简化了分离回收的操作。
(4) 极性DMF溶液和甲苯溶液有利于Knoevenagel缩合反应的进行, 催化剂重复使用5次后仍然具有很好的活性。由于复合材料中MOF组分的存在, 催化剂出现了一定程度的团聚, 这也是影响材料的比表面积的一个重要因素。因此, 还需进一步改进催化剂制备工艺, 使NMM-2催化剂具有更加优异的工业应用价值。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|