


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
刚玉型复合氧化物催化剂Cr1.3Fe0.7O3上二氯甲烷催化燃烧
[宁想1 , 张婷婷1 , 邢金媛1 , 陈家喜1 , 李艳明2 , 鲁继青1, *  ]
]
]
|
|


作者简介:宁 想,1992年生,女,河南省平顶山市人,在读硕士研究生。
通过溶胶-凝胶法合成一系列具有刚玉结构的Cr1.3Fe0.7O3复合氧化物,在二氯甲烷催化燃烧反应中进行活性评价。结果表明,复合氧化物比单金属氧化物CrOx和FeOx具有更高的催化活性,其催化活性与氧化物的表面酸性和可还原性密切相关,二者在反应中协同作用。与高温焙烧样品相比,低温焙烧的Cr1.3Fe0.7O3催化剂具有更高的比表面积、表面酸性和可还原性,催化性能更优。以表面酸性位数量为基础计算的催化剂转化频率(TOF)表明,各Cr1.3Fe0.7O3催化剂活性相近(300 ℃时约2.3×10-3 s-1~2.8×10-3 s-1),表面酸性位上发生二氯甲烷分子吸附及C-Cl键断裂,是反应活性位。300 ℃时Cr6+的TOF为9.3×10-3 s-1,Cr3+的TOF为0.59×10-3 s-1,Cr6+物种比Cr3+物种具有更高的活性。
A series of Cr1.3Fe0.7O3composite oxides with corundum structures were synthesized by sol-gel method and tested in catalytic combustion of dichloromethane (DCM). These oxides were much more active than monometallic CrO x and FeO xoxides.The catalytic performance was closely related to surface acidity and reducibility of the oxide,which showed clear synergistic effect in the reaction.Compared to those calcined at higher temperature,Cr1.3Fe0.7O3catalysts calcined at lower temperature had higher surface area,surface acidity and reducibility which were responsible for better catalytic performance.The similar average turnover frequencies (TOFs) for all the catalysts based on surface acidities (about 2.3×10-3s-1-2.8´10-3 s-1at 300 ℃) suggested that surface acid sites were active sites for the reaction since they provided centers for DCM chemisorption and subsequent cleavage of C-Cl bond.Moreover,the calculation revealed that the high-valent Cr6+ species were more active than the Cr3+ (with a TOF of 9.3×10-3 s-1versus 0.59´10-3 s-1at 300 ℃).
随着化工行业的发展和人类活动的增多, 空气污染已成为非常严重且越来越被关注的问题[1, 2, 3, 4]。挥发性有机化合物(VOCs), 尤其是氯化挥发性化合物(CVOCs)是最重要的工业污染物之一[5, 6]。CVOCs如1, 2-二氯乙烷(DCE), 二氯甲烷(DCM)和三氯乙烯(TCE)毒性高、难处理、破坏臭氧层、生物降解性低, 被认为是造成空气污染的主要物质。在CVOCs减排的各种技术中, 低温催化燃烧被认为是最有效和最有前途的技术之一[7, 8, 9, 10, 11]。
含Cr氧化物是用于CVOCs催化燃烧的最有效的过渡金属氧化物之一[12, 13, 14]。Su等[15]报道, 不同Cr含量(质量分数1%~20%)的Cr/HZSM-5催化剂对于DCM催化燃烧非常有效。Ma等[16]采用不同Cr含量的CrOx/Al2O3催化剂进行DCM氧化测试, 在350 ℃时, 质量分数为18%的Cr催化剂能够完全催化氧化DCM, 具有最佳的催化性能。在反应过程中, CrOx氧化物催化剂常发生活性Cr物种的流失, 导致催化剂严重失活。避免Cr物种流失的一种方法是将Cr限域在固定的晶型结构中。含Cr尖晶石型氧化物(ACr2O4)中Cr物种被限制在刚性晶格中, 避免Cr流失, 保持催化剂稳定性。据文献[17, 18]和我们的前期工作[19, 20, 21, 22, 23, 24], 含铬尖晶石氧化物是高活性和高稳定性的CVOCs催化燃烧催化剂。
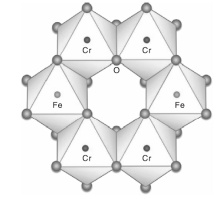
Cr1.3Fe0.7O3复合氧化物结构如图1所示。
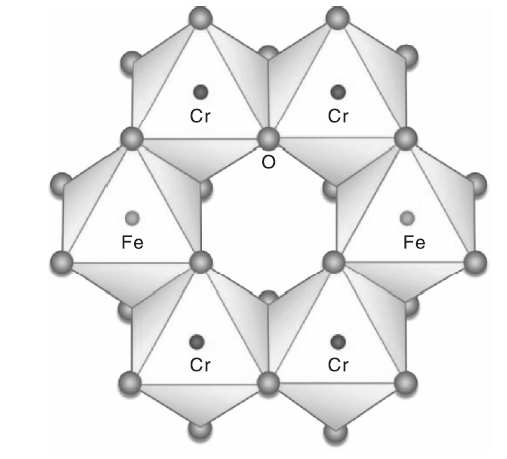 | 图1 Cr1.3Fe0.7O3的晶体结构图Figure 1 Crystal structure of Cr1.3Fe0.7O3 |
Cr1.3Fe0.7O3复合氧化物具有刚玉结构, 氧离子沿垂直立方轴六面最紧密排布。每个Cr3+或Fe3+阳离子被六个氧原子包围, 同时氧被[CrO6]八面体和[FeO6]八面体包围[25, 26, 27, 28]。这种Cr1.3Fe0.7O3氧化物广泛用作颜料[29]、磁电晶体[30]、陶瓷纳米颜料[31]和高效光催化剂[32]。在这样的氧化物晶格中, Cr离子暴露在表面并固定在晶格中, 该化合物可能是潜在的CVOCs氧化催化剂。
本文通过溶胶-凝胶法合成一系列Cr1.3Fe0.7O3复合氧化物催化剂, 进行DCM催化燃烧测试, 研究Cr1.3Fe0.7O3复合氧化物结构特性与催化性能间的关系, 讨论表面酸性和可还原性对催化性能的影响。
称取Fe(NO3)3· 9H2O 2.83 g、Cr(NO3)3· 9H2O 5.20 g和柠檬酸8.40 g, 物质的量比为0.7: 1.3: 6, 混合后加入100 mL去离子水, 50 ℃水浴搅拌30 min溶解, 90 ℃下持续搅拌4 h形成褐色凝胶。室温冷却, 120 ℃干燥过夜, (400~900) ℃空气气氛中焙烧4 h, 升温速率10 ℃· min-1, 制得催化剂Cr1.3Fe0.7O3-x(x=4、5、6、7和9)粉末, 其中x为温度参数。压片, 过筛, 取(40~60)目催化剂颗粒。纯FeOx和CrOx氧化物以类似的方式制备, 400 ℃下焙烧4 h, 命名为FeOx-4和CrOx-4。所需试剂均购自国药集团化学试剂有限公司, 无需进一步纯化。
采用Quanta chrome Autosorb-1型N2物理吸附仪测定催化剂比表面积, BET法计算, 液氮温度77 K下吸附氮气, 120 ℃真空预处理6 h。
采用Bruker D8 ADVANCE粉末X射线衍射仪进行催化剂的XRD表征, CuKα , 工作电压40 kV, 工作电流40 mA, 扫描范围10° ~90° , 扫描速率0.15° · s-1。
采用Hitachi S-4800型扫描电子显微镜观测复合氧化物催化剂表面的形貌, 催化剂需喷金处理, 工作电压5.0 kV。
采用H2-TPR测定催化剂的还原性, 高纯N2流量30 mL· min-1、温度300 ℃条件下, 将30 mg催化剂预处理0.5 h除去吸附水和碳酸盐, 冷却至50 ℃, 切换至体积分数5%H2-N2混合气, 流量30 mL· min-1, 以 10 ℃· min-1的速率升温至860 ℃。采用气相色谱热导池检测器测定氢气浓度变化, CuO样品标定氢气消耗量。
采用NH3-TPD测定催化剂的表面酸性, 高纯N2流量30 mL· min-1、温度300 ℃条件下, 预处理300 mg催化剂, 冷却至50 ℃, 通入30 mL· min-1的NH3吸附0.5 h。N2吹扫0.5 h, 升温至80 ℃, 继续通入30 mL· min-1的N2除去物理吸附的NH3。以10 ℃· min-1的速率由80 ℃升温至840 ℃, 采用气相色谱热导池检测器测定升温过程中NH3变化。
采用ESCALAB 250Xi型仪器进行催化剂的X射线光电子能谱测定。AlKa, 工作电压20 kV, 真空度约2× 10-7Pa, 通过C ls(284.6 eV)校正结合能。
催化剂活性评价在微型固定床反应器上进行。称取1 g催化剂与相同粒径的石英砂均匀混合至2 mL, 装入内径为9 mm石英反应管。反应气中二氯甲烷物质的量浓度为0.3%, 水蒸气物质的量浓度为1.2%, 其余为空气, 气体总流量500 mL· min-1, 空速15 000 h-1。反应尾气首先通过0.1 mol· L-1的NaOH吸收液除去反应中生成的氯气或氯化氢, 再由配备氢火焰离子检测器(FID)的气相色谱测定尾气浓度。根据反应前后气体峰面积的百分比计算二氯甲烷的转化率。
反应动力学研究在连续流动固定床反应器上进行。实验中采用微分模式, 保证DCM转化率小于15%。实验步骤与反应条件与催化剂性能测试相同。通过Weisz-Prater和Mears标准的评估, 排除动力学研究过程中传质和传热的影响。
Cr1.3Fe0.7O3催化剂的SEM照片如图2所示, 由图2可知所有样品均为不规则颗粒状且无孔道结构。
 | 图2 不同Cr1.3Fe0.7O3催化剂的SEM照片Figure 2 SEM images of various Cr1.3Fe0.7O3 catalysts |
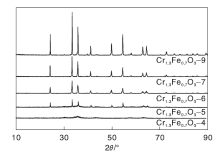
图3为Cr1.3Fe0.7O3催化剂的XRD图。
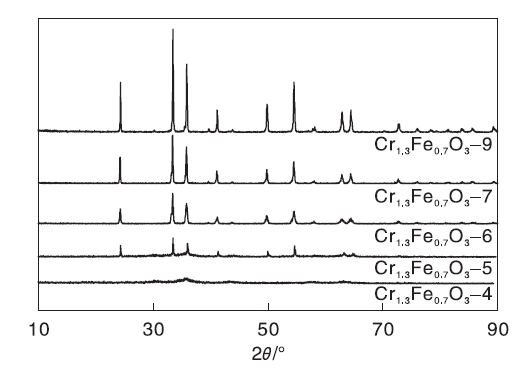 | 图3 Cr1.3Fe0.7O3 催化剂的XRD图Figure 3 XRD patterns of Cr1.3Fe0.7O3catalysts |
由图3可知, 400 ℃焙烧的样品Cr1.3Fe0.7O3-4的衍射峰非常弱且宽, 表明该样品是无定形结构。样品焙烧温度高于500 ℃时, 在2θ 值为24.3° 、33.4° 、35.9° 、41.1° 、49.8° 、54.55° 、58.1° 、62.9° 和64.4° 处观察到明显的纯刚玉特征衍射峰(JCPDS No.35-1112)。随着焙烧温度的升高, 衍射峰强度持续增加, 表明烧结效应导致晶粒尺寸的增大。
基于XRD数据, 采用谢乐(Scherrer)方程计算Cr1.3Fe0.7O3氧化物的晶粒尺寸和晶胞参数, 结果如表1所示。由表1可知, 催化剂的晶粒尺寸随着焙烧温度的升高逐渐增大, 由Cr1.3Fe0.7O3-5的17.2 nm增加至Cr1.3Fe0.7O3-9的55.4 nm。高温烧结导致催化剂比表面积逐渐下降。 Cr1.3Fe0.7O3-4样品的比表面积高达169.4 m2· g-1, 比Cr1.3Fe0.7O3-9的3.3 m2· g-1高50倍。
| 表1 催化剂的比表面积和晶粒尺寸 Table 1 Specific surface areas and particle sizes of catalysts |
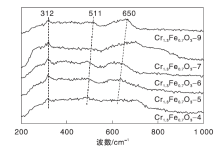
Cr1.3Fe0.7O3催化剂的拉曼光谱图如图4所示。由图4可知, 312 cm-1、511 cm-1和650 cm-1处出现谱带, 归属于刚玉型Cr1.3Fe0.7O3氧化物的特征峰[32]。
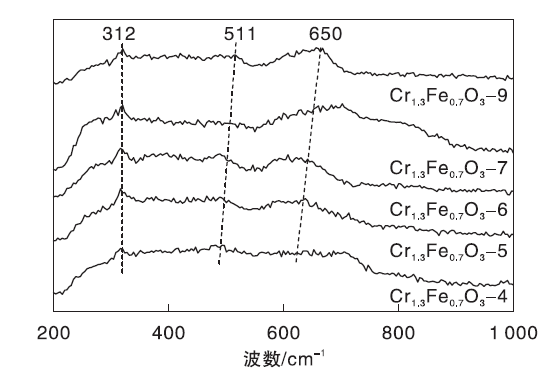 | 图4 Cr1.3Fe0.7O3 催化剂的拉曼光谱图Figure 4 Raman spectraof Cr1.3Fe0.7O3catalysts |
图5是Cr1.3Fe0.7O3催化剂的Cr 2p、Fe 2p和O 1s XPS谱图。由图5可知, Cr 2p3/2谱图可拟合为位于575.9 eV、577.1 eV和578.8 eV的3个峰, 分别归属于Cr(OH)3或Cr2O3、Cr3+(占据八面体位置)和Cr6+[33, 34]。Fe 2p3/2谱图中结合能为710.3 eV的峰归属于Fe2+, 结合能为711.6 eV、713.4 eV和715.3 eV的峰归属于Fe3+, 结合能719.1 eV的峰归属于Fe3+的卫星峰[35, 36, 37]。O 1s光谱图中530.2 eV和531.3 eV处的峰分别归属于吸附的氧物种(Oads, 化学吸附的氧, 表面羟基和表面H2O等)和晶格氧物种(Olatt)[24]。
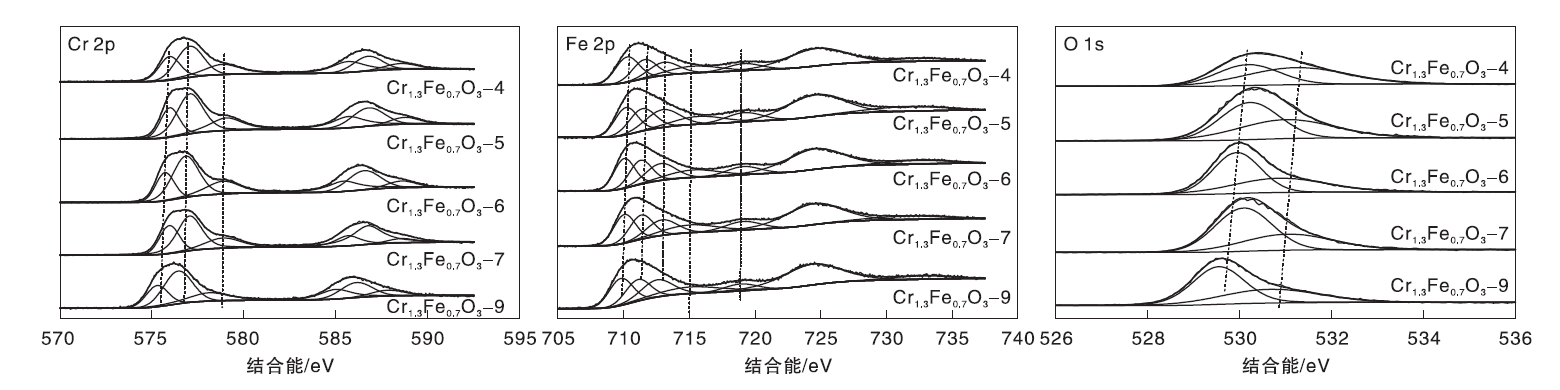 | 图5 Cr1.3Fe0.7O3催化剂的XPS谱图Figure 5 XPS spectra of Cr1.3Fe0.7O3catalysts |
表2是Cr1.3Fe0.7O3催化剂的表面组成。
| 表2 Cr1.3Fe0.7O3催化剂的表面组成 Table 2 Surface composition of Cr1.3Fe0.7O3catalysts |
由表2可知, Cr1.3Fe0.7O3催化剂中表面Fe3+阳离子含量约70%, Cr6+含量20%~26% (对应的Cr3+含量为74%~80%)。低温焙烧的样品含有较高浓度的表面吸附氧物种。Cr1.3Fe0.7O3-4的Oads/Otot物质的量比为0.56, Cr1.3Fe0.7O3-9的Oads/Otot物质的量比为0.36。这是由于无定型结构的
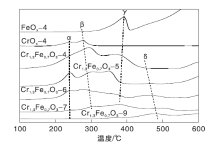
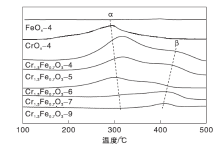
催化剂的H2-TPR曲线如图6所示。由图6可知, FeOx-4样品在约400 ℃处出现还原峰γ , 这是由于Fe2O3还原成Fe3O4。CrOx-4在(200~300) ℃有重叠的还原峰, 可归属于CrO3还原成Cr2
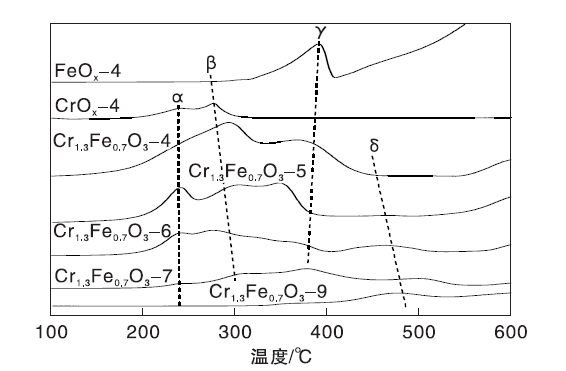 | 图6 Cr1.3Fe0.7O3催化剂的H2-TPR曲线Figure 6 H2-TPR profiles of Cr1.3Fe0.7O3catalysts |
催化剂的耗H2量如表3所示。由表3可知, Cr1.3Fe0.7O3-4的耗H2量为1.98 mmol· g-1, Cr1.3Fe0.7O3-9的耗H2量降至0.10 mmol· g-1, 这可能是由于高温焙烧使比表面积下降。另一方面, 归属于Cr6+物质还原的峰α 随着焙烧温度的增加而降低, 表明高温下焙烧的氧化物中Cr6+物种的量下降。
| 表3 催化剂的耗氢量和表面酸量 Table 3 H2 consumption and surface acidity of catalysts |
催化剂的NH3-TPD曲线如图7所示。由图7可知, FeOx-4样品几乎无表面酸性, CrOx-4样品在(200~400) ℃有宽的NH3脱附峰α (集中在300 ℃), 表明CrOx-4中存在中等强度的Lewis酸[41]。Cr1.3Fe0.7O3氧化物在300 ℃有强NH3脱附峰α , 在400 ℃有弱NH3脱附峰β 。前者归属于中等强度的Lewis酸位, 后者为强Lewis酸位。随着焙烧温度的升高, 峰α 、峰β 强度下降。
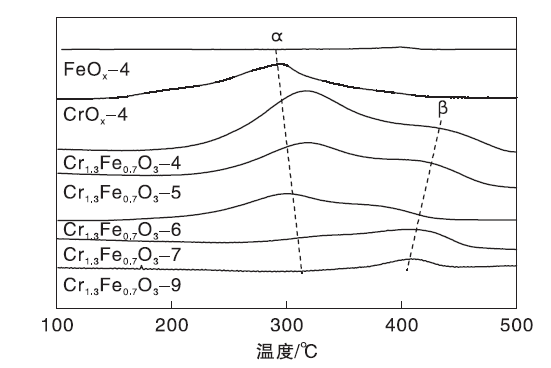 | 图7 Cr1.3Fe0.7O3 催化剂的NH3-TPD曲线Figure 7 NH3-TPD profiles of Cr1.3Fe0.7O3catalysts |
催化剂的表面酸量如表3所示。Cr1.3Fe0.7O3-4表面酸量最大为0.42 mmol· g-1, Cr1.3Fe0.7O3-9表面酸量最低为0.04 mmol· g-1。
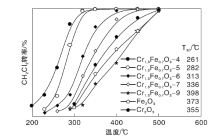
Cr1.3Fe0.7O3催化剂的DCM催化燃烧活性如图8所示。
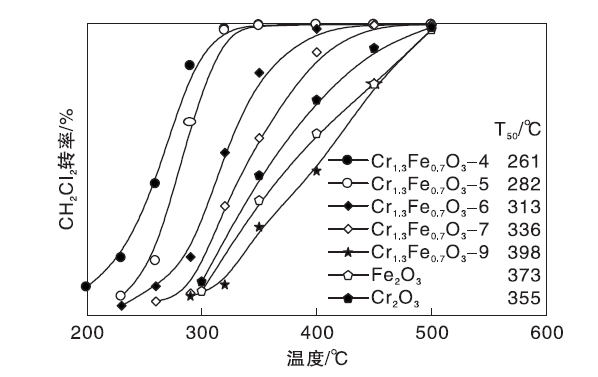 | 图8 Cr1.3Fe0.7O3催化剂的DCM催化燃烧活性图Figure 8 Catalytic combustion activity of DCM on Cr1.3Fe0.7O3catalysts |
由图8可知, 活性顺序为Cr1.3Fe0.7O3-9﹤Fe2O3﹤Cr2O3﹤Cr1.3Fe0.7O3-7﹤Cr1.3Fe0.7O3-6﹤Cr1.3Fe0.7O3-5﹤Cr1.3Fe0.7O3-4, T50(转化率为50%时的温度)值分别为398 ℃、373 ℃、355 ℃、336 ℃、313 ℃、282 ℃和261 ℃。
Cr1.3Fe0.7O3-4比单组分的CrOx-4和FeOx-4活性更强, 表明复合氧化物在DCM催化燃烧反应中具有优势。反应温度为300 ℃时, Cr1.3Fe0.7O3-4、CrOx-4和FeOx-4催化剂的质量比速率分别为0.99 μ mol· g-1· s-1、0.13 μ mol· g-1· s-1和0.09 μ mol· g-1· s-1。对比Cr1.3Fe0.7O3-4、CrOx-4和FeOx-4催化剂的面积比速率0.584´ 10-8 mol· m-2· s-1、0.179´ 10-8 mol· m-2· s-1和0.187´ 10-8 mol· m-2· s-1, 也验证了该结论。
随着焙烧温度的升高, Cr1.3Fe0.7O3-x催化剂的活性逐渐降低。Cr1.3Fe0.7O3-4催化剂(250 ℃时1.61 mmolCH2Cl2·
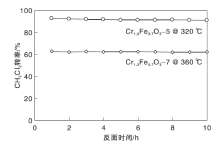
Cr1.3Fe0.7O3-5和Cr1.3Fe0.7O3-7催化剂的稳定性如图9所示。由图9可知, Cr1.3Fe0.7O3-5和Cr1.3Fe0.7O3-7催化剂在10 h反应时间内稳定性良好, 表明反应过程中未发生Cr物种流失或表面积炭。
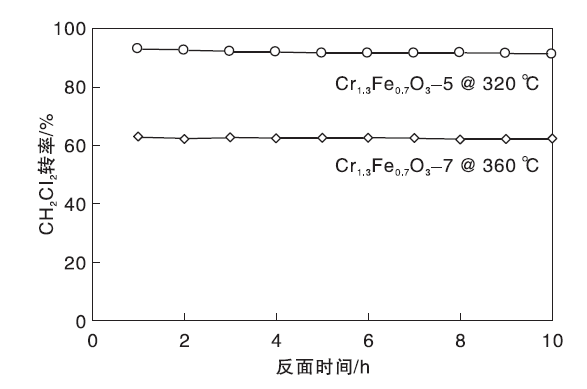 | 图9 Cr1.3Fe0.7O3-5和Cr1.3Fe0.7O3-7催化剂的稳定性Figure 9 Stability of Cr1.3Fe0.7O3-5and Cr1.3Fe0.7O3-7catalysts |
对于CVOCs催化氧化, 表面酸性和催化剂还原性是至关重要的两个参数, 这是由于表面酸性位点为CVOCs分子提供化学吸附中心, 催化剂的还原性则影响反应中氧物种的迁移和活化, 通常两者协同作用影响催化剂的催化行为[2, 5, 19, 21, 24]。本文中, NH3-TPD和H2-TPR结果表明Cr1.3Fe0.7O3-4具有最高的表面酸性(0.43 mmol· g-1)和最强的可还原性(1.98 mmol· g-1), 这很好地解释了Cr1.3Fe0.7O3-4具有最佳的催化反应活性。
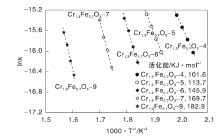
动力学研究也证明低温焙烧的催化剂比高温焙烧的催化剂活性更高。图10为Cr1.3Fe0.7O3催化剂的CH2Cl2氧化的Arrhenius图。由图10可知, 在DCM转化率小于15%的微分反应条件下, 表观活化能(Eas)大小为Cr1.3Fe0.7O3-4(101.6 kJ· mol-1)﹤Cr1.3Fe0.7O3-5(113.7 kJ· mol-1)﹤Cr1.3Fe0.7O3-6(145.9 kJ· mol-1)﹤Cr1.3Fe0.7O3-7(169.7 kJ· mol-1)﹤Cr1.3Fe0.7O3-9(182.9 kJ· mol-1), 与反应活性顺序一致。
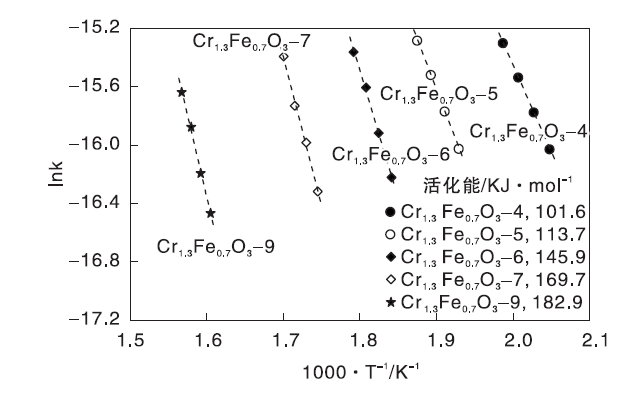 | 图10 Cr1.3Fe0.7O3催化剂的CH2Cl2氧化的Arrhenius图Figure 10 Arrhenius plots of oxidation of CH2Cl2over Cr1.3Fe0.7O3 catalysts |
图11为反应速率与催化剂还原性/表面酸性之间的关系。由图11可以看出, 反应速率与表面酸性和催化剂还原性成正比。
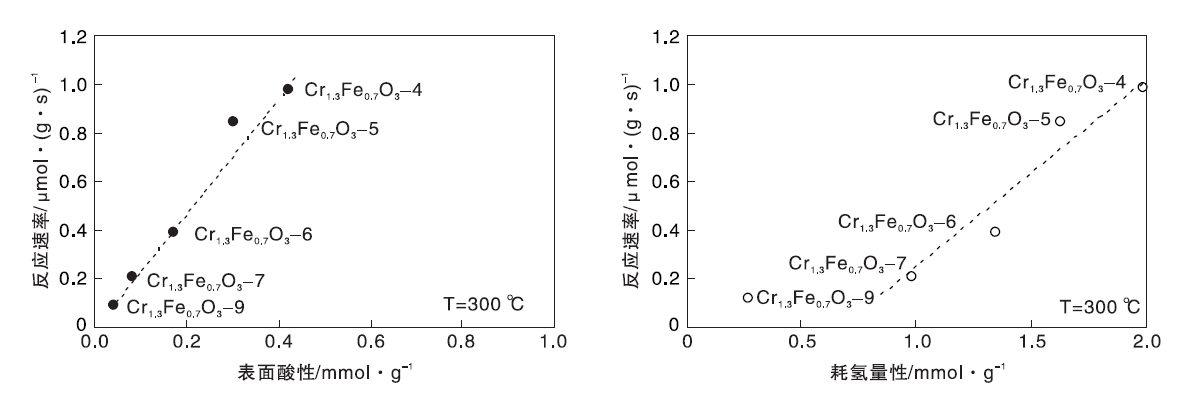 | 图11 反应速率与催化剂还原性/表面酸性之间的关系Figure 11 Relationship between reaction rate and catalyst reduction/surface acidity |
表面酸量可能与催化剂中Cr物种的存在有关, 由于Cr3+的极性比Fe3+大, 导致表面酸性增加。按照以下公式估算表面Cr离子的量(mol· g-1):
式中,
Cr1.3Fe0.7O3-4、Cr1.3Fe0.7O3-5、Cr1.3Fe0.7O3-6、Cr1.3Fe0.7O3-7和Cr1.3Fe0.7O3-9表面Cr离子含量计算值分别为797.8 μ mol· g-1、187.9 μ mol· g-1、51.3 μ mol· g-1、33.0 μ mol· g-1和15.5 μ mol· g-1。由于非晶特性, Cr1.3Fe0.7O3-4的计算值误差可能较大。
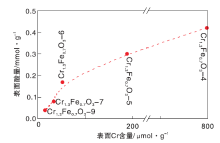
催化剂表面酸量与Cr含量的关系如图12所示。由图12可知, 表面酸量随着Cr含量的增加而增加, 表明Cr离子对表面酸性起主要作用。这与NH3-TPD结果一致。
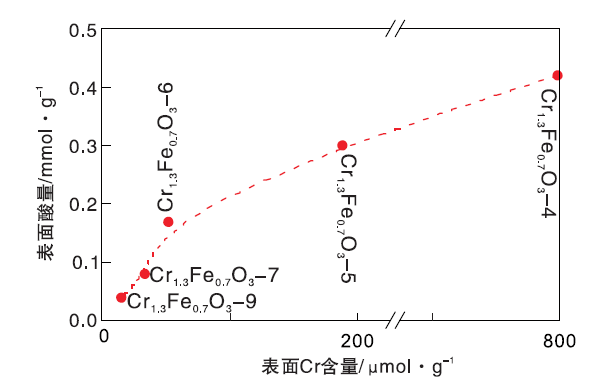 | 图12 催化剂表面酸量与Cr含量的关系Figure 12 Relationship between catalyst surface acidity and Cr content |
反应温度为300 ℃时, Cr1.3Fe0.7O3催化剂的面积比速率和转化频率如表4所示。
| 表4 Cr1.3Fe0.7O3催化剂的面积比速率和转化频率 Table 4 Areal reaction rate and turnover frequency of Cr1.3Fe0.7O3catalysts |
由表4可以看出, Cr1.3Fe0.7O3-6催化剂的面积比速率值最大为3.62× 10-8 mol· m-2· s-1。基于表面酸量的转化频率[TOF, TOF=质量比反应速率(mol· g-1· s-1)/表面酸量(mol· g-1)]值相近(2.23~2.83)´ 10-3 s-1, 表明表面酸性位点可能是反应活性位, 表面酸性位点与DCM分子的吸附和C-Cl键的断裂[45]有关。
据我们以前的工作[21], 对于尖晶石CoCr2O4氧化物催化剂, 高价Cr6+比低价Cr3+活性更强。因此, 基于下式计算不同阳离子的TOF:
TOFavarage = TO
式中,
由于FeOx-4在300 ℃时基本无活性, 公式简化为:
TOFavarage = TO
采用表4和表2中的数据并进行多元线性回归, 计算得到Cr6+和Cr3+在300 ℃的TOF分别为9.3× 10-3 s-1和0.59× 10-3 s-1, Cr6+的TOF比Cr3+高15倍, 与文献[21]一致。与Cr3+相比, Cr6+ TOF的增大可能是由于可还原性及表面酸性增强, 这是催化剂活性的两个决定性因素。
(1)刚玉结构的Cr1.3Fe0.7O3复合氧化物具有较高的DCM催化燃烧活性, 其催化活性与表面酸性和还原性协同作用相关。
(2)与高温焙烧样品相比, 低温焙烧的Cr1.3Fe0.7O3催化剂具有更高的比表面积、表面酸性和可还原性, 具有更高的催化性能。
(3)以表面酸性位数量为基础计算的催化剂转化频率表明, Cr1.3Fe0.7O3催化剂具有相近的活性, 300 ℃时, TOF约(2.3~2.8)× 10-3 s-1, 表明表面酸性位是反应活性位, 表面酸性位是二氯甲烷分子吸附及C-Cl键断裂的活性位。高价Cr6+比Cr3+具有更高的活性, 300 ℃时, Cr6+的TOF为9.3× 10-3 s-1, Cr3+的TOF为0.59 × 10-3 s-1。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|