

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
添加WO3对铈钴催化剂CO氧化性能的影响
引用本文
史蕊, 李坚. 添加WO3对铈钴催化剂CO氧化性能的影响 [J]. 工业催化, 2018,26(3): 39-44.
Shi Rui, Li Jian. Effect of addition WO3 to cerium-cobalt catalysts on carbon monoxide catalytic oxidation performance [J]. Industrial Catalysis, 2018,26(3): 39-44.
DOI:10.3969/j.issn.1008-1143.2018.03.008
Shi Rui, Li Jian. Effect of addition WO3 to cerium-cobalt catalysts on carbon monoxide catalytic oxidation performance [J]. Industrial Catalysis, 2018,26(3): 39-44.
Permissions
Copyright©2018, 《工业催化》编辑部
《工业催化》编辑部 所有
添加WO3对铈钴催化剂CO氧化性能的影响
作者简介:史 蕊,1993年生,女,北京市人,在读硕士研究生。
摘要
采用共沉淀法制备 xWO3-CeO2-Co3O4复合型非贵金属CO低温催化剂,考察不同WO3添加量和空速对催化剂催化活性的影响,并考察催化剂的抗硫性能。通过孔隙结构测试、H2-TPR、FT-IR和SEM等对催化剂进行表征。结果表明,WO3添加质量分数1%时,催化剂具有最佳的低温活性。在CO进口体积分数0.12%、O2进口体积分数5%和空速15 000 h-1条件下,50 ℃时,CO转化率即可达到99.6%,60 ℃时,CO转化率达100%。添加WO3,催化剂氧化能力增强,催化效率提高。随着空速升高,CO转化率下降。WO3的加入可有效提高催化剂的比表面积,抑制硫酸盐在催化剂表面聚集,提高催化剂的抗硫性能。
关键词:
催化化学; CO催化氧化; CO低温催化剂; WO3; Co3O4; 抗硫性能
中图分类号:O643.36;X51
文献标志码:A
文章编号:1008-1143(2018)03-0039-06
Effect of addition WO3 to cerium-cobalt catalysts on carbon monoxide catalytic oxidation performance
Abstract
xWO3-CeO2-Co3O4 catalysts for low temperature oxidation of carbon monoxide were prepared by coprecipitation method and characterized by pore structure test,H2-TPR,FT-IR,SEM.The effects of WO3content and space velocity on activity were investigated.Sulphur resistance of catalyst was also studied.The research showed that the catalyst with 1% WO3 addition had the best activity for low-temperature CO catalytic oxidation.The CO conversion could reach 99.6% at 50 ℃ and 100% at 60 ℃ under the condition of inlet CO concentration of 0.12%,O2 volume fraction of 10%,and space velocity of 15 000 h-1.Adding WO3 to CeO2-Co3O4could increase catalysts oxidation ability and catalyst efficiency.CO conversion decreased with increasing space velocity.The addition of WO3 could effectively improve the specific surface area of catalyst,inhibit aggregation of sulfate on catalysts surface and improve sulfur resistance of the catalysts.
Keyword:
catalytic chemistry; catalytic oxidation of carbon monoxide; low temperature oxidation catalyst of CO; WO3; Co3O4; sulfur resistance
CO是污染空气、威胁人类和动物健康的气体[1, 2], 因无色无味所以在日常生活中很容易忽视而存在危险。产生CO的途径有很多, 比如汽车尾气、香烟烟雾、煤气炉、木材炉、化石燃料不完全燃烧、采矿工业和发电厂等[3], 其中, 人为源主要来自汽车尾气与工业排放[4], 自然源主要是海洋、森林火灾和森林中释放的萜烯化合物以及其他生物体的燃烧等。对CO的排放控制已成为环境治理的重点, 受到广泛关注。
催化氧化法可以有效去除CO, 已应用在环境保护、能源、化工、材料和军工等方面。其中, 低温CO催化氧化可以用于密闭系统CO消除、消防自救呼吸器和地下矿井过滤自救器等[5]。CO低温催化氧化催化剂主要分为非贵金属催化剂和贵金属催化剂[6]。研究较多的贵金属催化剂包括金系、银系、铂系和钯系等。研究表明[7, 8], 贵金属催化剂催化性能良好, 能够在较低温度下取得较高的催化活性, 但在高温下易烧结, 价格昂贵, 易硫中毒, 因此在实际应用中受到限制。非贵金属催化剂价格相对低廉, 主要有铜系、钴系[9]和锰系等单组分非贵金属以及负载型和复合型的非贵金属催化剂。霍加拉特催化剂是由CuO和MnO2按照一定比例制备而成的非贵金属催化剂, 催化活性较好, 是目前应用最广泛也是最古老的CO氧化催化剂[10]。
本文在CeO2-Co3O4催化剂中添加WO3以实现更好的CO低温催化性能, 采用共沉淀法制备xWO3-CeO2-Co3O4复合型非贵金属CO低温催化剂, 考察WO3添加量、空速和SO2对催化剂CO催化性能的影响。
1 实验部分
1.1 仪器与试剂
乙酸钴、硝酸铈、钨酸铵(85.0%~90.0%)、无水碳酸钠、无水乙醇溶液, 均为分析纯, 天津市福晨化学试剂厂; CO, 8%、O2, 99.9%, 工业纯、N2, 99.9%, 工业纯、SO2, 2%, 北京市海瑞通达气体有限公司。
KQ2200E型超声波清洗器, 昆山市超声仪器有限公司; JJ-1精密定时电动搅拌器, 江苏省金坛市荣华仪器制造有限公司; D07-7B型质量流量控制器、D08-1F型流量显示仪, 北京七星华创电子股份有限公司; CKW-1100型温度控制器, 北京市朝阳自动化仪表厂; 德国德图公司testo 350 CO烟气分析仪, 量程0~+10 000× 10-6, 分辨率1× 10-6。
1.2 催化剂制备
采用共沉淀法制备不同WO3含量的xWO3-CeO2-Co3O4复合型非贵金属CO低温催化剂。将一定量乙酸钴、硝酸铈和钨酸铵充分搅拌溶解, 加入碳酸钠形成沉淀物, 70 ℃水浴条件下继续搅拌3 h, 倒入离心管中离心洗涤5次以去掉乙酸根、硝酸根和钠离子。将洗涤过的沉淀物放入烘箱, 110 ℃干燥12 h, 烘干后块状固体放入马弗炉400 ℃焙烧3 h。焙烧后催化剂冷却后进行研磨筛分, 制成(20~40)目颗粒备用。
1.3 催化剂活性评价
催化剂催化氧化CO实验在固定床玻璃反应管(ϕ 19 mm× 250 mm)中进行, 通过加热电炉控制反应管温度, 测试温度为(30~100) ℃。测试条件为:CO进口体积分数0.12%, O2进口体积分数5%, N2作为平衡气, 空速15 000 h-1, 抗硫实验时SO2体积分数为0.02%。采用CO烟气分析仪检测反应进出口CO浓度, 计算CO转化率。
1.4 催化剂表征
H2-TPR采用美国麦克仪器公司AutochemⅡ 2920化学吸附仪测试。催化剂在O2条件下500 ℃预处理60 min, 冷却至室温, 改用氦气吹扫15 min, 然后通入5%H2-Ar, 升温速率为10 ℃· min-1, 通过热导检测器(TCD)在线记录H2浓度。用CuO标样(纯度为99.995%)标定H2消耗量。
催化剂孔隙结构由美国麦克仪器公司Gemini Ⅴ 比表面积及孔隙度分析仪测定。催化剂在110 ℃预处理1 h, 比表面积按BET方法计算, 孔容和孔径按BJH理论计算。
FT-IR采用Nicolet 6700傅里叶红外光谱分析仪测试。将催化剂与烘干KBr按一定比例混合、研磨, 用压片机制成薄片, 傅里叶红外光谱分析仪扫描。
SEM采用日本日立公司JSM7610F型场发射电子显微镜测试, 对催化剂进行微观成像, 工作电压10 kV。
2 结果与讨论
2.1 WO3添加量
目前, 添加WO3的钒钨钛SCR催化剂工业应用较为广泛[11, 12]。采用共沉淀法制备WO3质量分数为0~6%的xWO3-CeO2-Co3O4催化剂, 在CO体积分数0.12%、O2体积分数5%、N2作为平衡气和空速15 000 h-1条件下, 考察WO3添加量对催化剂催化活性的影响, 结果见表1。
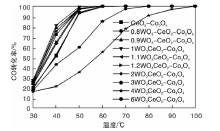
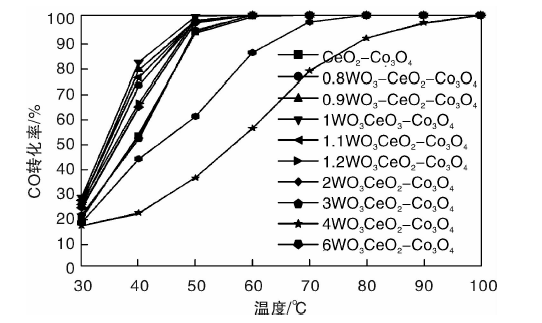 | 图1 WO3添加量对催化剂催化活性的影响Figure 1 Effect of WO3addition amount on the catalytic activity |
从图1可以看出, WO3添加质量分数1%时, 1WO3-CeO2-Co3O4催化剂对CO催化活性最好, 50 ℃时, CO转化率即可达99.6%, 60 ℃时, CO转化率100%。WO3添加质量分数超过1%, 催化活性随着WO3添加量的增加逐渐变差。未添加WO3时, CeO2-Co3O4催化剂在70 ℃时CO去除率100%, 添加1%WO3可使CO去除率100%的温度降低10 ℃。WO3具有一定的储存和转移电子的能力, 并且可以促进催化剂表面的超氧自由基数量增加, 提高CO催化剂催化氧化性能[13]。
对比市售霍加拉特催化剂与1WO3-CeO2-Co3O4催化剂的CO催化活性, 结果如图2所示。
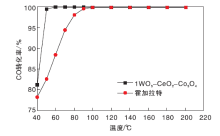
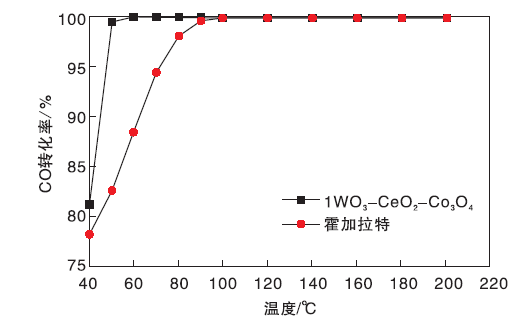 | 图2 1WO3-CeO2-Co3O4催化剂与霍加拉特催化剂活性对比Figure 2 Efficiency comparisons between 1WO3-CeO2-Co3O4catalyst and Hopcalite catalyst |
从图2可以看出, 1WO3-CeO2-Co3O4催化剂催化CO效果好于市售霍加拉特催化剂, 霍加拉特催化剂在100 ℃时CO去除率才可达到100%
2.2 H2-TPR
1WO3-CeO2-Co3O4催化剂与未添加钨CeO2-Co3O4催化剂进行H2-TPR表征, 结果如图3所示。
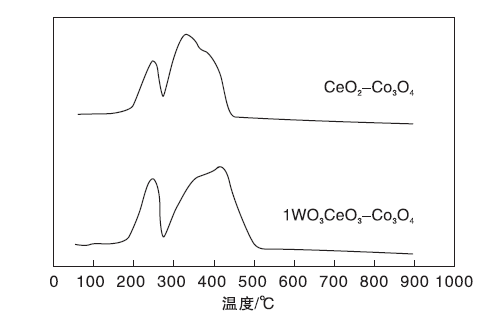 | 图3 CeO2-Co3O4和1WO3-CeO2-Co3O4催化剂的H2-TPR谱图Figure 3 H2-TPR profiles of CeO2-Co3O4 and 1WO3-CeO2-Co3O4 catalysts |
由图3可见, 两个催化剂均具有两个还原峰。CeO2-Co3O4催化剂的低温还原峰在249 ℃, 1WO3-CeO2-Co3O4催化剂的低温还原峰在243 ℃。通过计算得到CeO2-Co3O4催化剂低温耗氢量为8.46 mmol· g-1, 高温耗氢量为27.58 mmol· g-1; 1WO3-CeO2-Co3O4催化剂低温耗氢量为11.09 mmol· g-1, 高温耗氢量为33.71mmol· g-1, 由此可见, 添加钨的1WO3-CeO2-Co3O4催化剂具备更强的氧化还原能力, 可以促进CO催化氧化反应, 与低温CO催化活性测试结果相符。
2.3 催化剂结构参数
表1为不同WO3添加量xWO3-CeO2-Co3O4催化剂的结构参数。
| 表1 xWO3-CeO2-Co3O4催化剂结构参数 Table 1 Textural parameters of xWO3-CeO2-Co3O4 catalysts |
从表1可以看出, 随着WO3添加量的增加, 催化剂比表面积逐渐增大, 孔径逐渐减小, WO3添加质量分数1%时, 催化剂比表面积增幅最大, 孔径降幅最明显。添加WO3对催化剂孔容影响不大。较大比表面积使催化剂与CO和O2气体分子接触面积增加, 促进了催化氧化性能, 有利于催化剂低温活性提高; 催化剂孔径并不是越大越有利于提高催化活性, 适当孔径有利于气体分子进入。
2.4 空 速
在CO体积分数0.12%、O2体积分数5%和N2作平衡气条件下, 考察空速对1WO3-CeO2-Co3O4催化剂催化效率的影响, 结果如图4所示。
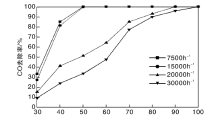
 | 图4 空速对CO去除率的影响Figure 4 Effect of space velocity on CO removal efficiency |
从图4可以看出, 随着空速的提高, CO去除率逐渐降低。空速7 500 h-1, CO完全转化温度为50 ℃; 空速30 000 h-1, CO完全转化温度为100 ℃。根据Langmuir-Hinshelwood机理[14], 空速提高, CO与O2接触时间变短, 气体与催化剂活性位点得不到充分接触, 使高空速下催化剂活性下降。
2.5 SO2对催化剂活性的影响
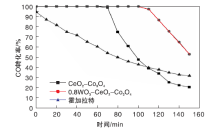
在温度70 ℃、CO体积分数0.12%、O2体积分数5%、SO2体积分数0.02%、N2作平衡气和空速15 000 h-1条件下, 选择低温催化活性最好的1WO3-CeO2-Co3O4催化剂与未添加WO3的CeO2-Co3O4催化剂及霍加拉特催化剂进行抗硫对比实验, 结果如图5所示。从图5可以看出, 1WO3-CeO2-Co3O4催化剂和CeO2-Co3O4催化剂60 min内催化活性未下降, CO去除率一直稳定在100%。反应70 min时, CeO2-Co3O4催化剂上CO去除率开始下降, 通硫120 min时, CO去除率下降至34.1%, 150 min时, CO去除率下降至20.8%; 1WO3-CeO2-Co3O4催化剂在100 min内催化活性均保持在100%, 120 min时, CO去除率下降至86.6%, 150 min时, CO去除率为53%。表明添加WO3有助于催化剂抗硫, 抑制硫酸盐在催化剂表面的形成。从图5还可以看出, 霍加拉特催化剂几乎没有抗硫性能。
 | 图5 SO2对催化剂活性的影响Figure 5 Effect of SO2on catalysts activity |
2.6 FT-IR
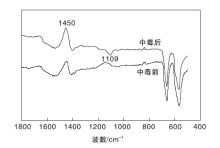
对SO2中毒前后1WO3-CeO2-Co3O4催化剂进行红外测试, 结果见图6。
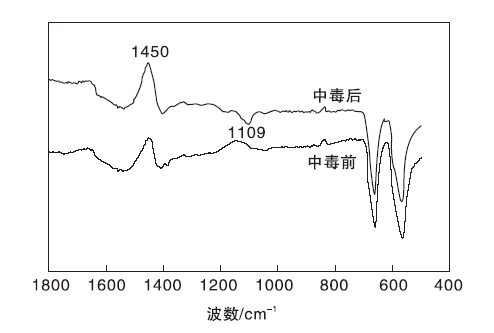 | 图6 SO2中毒前后1WO3-CeO2-Co3O4催化剂的FT-IR谱图Figure 6 FT-IR spectra of catalyst before and after poisoning |
从图6可以看出, SO2中毒前后1WO3-CeO2-Co3O4催化剂在565 cm-1、660 cm-1和1 450 cm-1出现了明显的特征峰, 其中, 565 cm-1和660 cm-1处两个尖锐吸收峰分别对应尖晶石结构四氧化三钴的Co(Ⅱ )-O(Ⅱ )和Co(Ⅲ )-O(Ⅰ )特征振动峰[15]。C
2.7 SO2中毒前后催化剂结构性能
表2为SO2中毒前后1WO3-CeO2-Co3O4和CeO2-Co3O4催化剂的结构性能。
| 表2 SO2中毒前后1WO3-CeO2-Co3O4和CeO2-Co3O4催化剂的结构性能 Table 2 Pore structure parameters of catalysts before and after poisoning |
从表2可以看出, SO2中毒催化剂的比表面积和孔容降低, 孔径提高。是因为通SO2后, 在催化剂表面形成了硫酸盐, 覆盖在催化剂表面, 堵塞孔道, 导致比表面积和孔容均下降。孔径提高是因为硫酸盐呈酸性对催化剂产生了扩孔作用。中毒前后CeO2-Co3O4催化剂变化更明显, 表明在抗硫实验过程中, 催化剂孔道不断被硫酸盐堵塞, 添加WO3可以起抑制硫酸盐沉积在催化剂表面的作用。
2.8 SEM
SO2中毒前后1WO3-CeO2-Co3O4催化剂的SEM照片见图7。从图7可以看出, 新鲜催化剂孔道清晰可见, 中毒催化剂表面看不到同样的孔状结构, 而是被团状物包覆, 呈现团聚现象。在通SO2实验过程中, 催化剂的孔道不断被硫酸盐堵塞, 导致催化剂催化活性降低。
 | 图7 SO2中毒前后1WO3-CeO2-Co3O4催化剂的SEM照片Figure 7 SEM photos of catalyst before and after poisoning |
3 结 论
采用共沉淀法制备xWO3-CeO2-Co3O4复合型非贵金属CO低温催化剂, WO3添加质量分数1%时, 催化剂具有最佳的CO低温催化氧化活性, 50 ℃时, CO转化率即可达到99.6%, 60 ℃时, CO转化率达100%。添加WO3有利于催化剂抗硫, 可以有效抑制硫酸盐在催化剂表面的形成。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|