{kind=link}
耐硫甲烷合成反应机理研究进展
[史立杰*  , 张永泽, 李晨佳, 冯璐瑶, 张金舵, 刘鹏翔, 常俊石]
, 张永泽, 李晨佳, 冯璐瑶, 张金舵, 刘鹏翔, 常俊石]
, 张永泽, 李晨佳, 冯璐瑶, 张金舵, 刘鹏翔, 常俊石]
|
|


作者简介:史立杰, 1979年生, 男,高级工程师,主要从事催化剂开发与工艺设计研究。
近年来,Mo基耐硫甲烷合成技术受到广泛关注。综述Mo基催化剂活性相的结构模型,主要包括Rim-Edge模型、协同作用模型、嵌入模型、单层结构模型、Co-Mo-S模型和双金属硫化物模型,其中Co-Mo-S模型的接受度最广。通过分析耐硫甲烷合成机理发现,反应体系中同时进行甲烷合成反应与水汽变换反应。
In recent years,sulfur-resistant methanation on Mo-based catalyst has received the widespread attention.The structure models of active phases of Mo-based catalysts are reviewed,including Rim-Edge model,contact synergy model,embedding model,single-layer structure model,Co-Mo-S model and bimetallic sulfide model.Among them,Co-Mo-S model has the widest acceptance.By analyzing the synthesis mechanism of sulfur-tolerant methanation,it is found that methane synthesis and water-gas shift reactions are carried out simultaneously in the reaction system.
甲烷合成是煤制天然气工艺的核心技术之一。传统甲烷合成工艺中, 主要采用Ni基催化剂, 已在工业中得到广泛应用[1]。传统甲烷合成工艺具有处理量大、工艺路线相对成熟、甲烷产率高等优点。但由于Ni基催化剂对原料气中的硫极为敏感, 一般要求原料气中硫含量低于0.1× 10-6。为防止催化剂积炭, 需经水汽变换反应将原料气的氢碳物质的量比调整至3以上。因此, 开发在低氢碳比且含硫气氛中仍具有较好活性的催化剂是甲烷合成技术的发展方向。
近年来, 耐硫甲烷合成技术成为研究的热点。由于该工艺所用原料气无须脱硫, 通常催化剂具有甲烷化与变换双重功能[2], 降低了该工艺对原料气氢碳比的要求, 极大简化了工艺流程, 达到节能降耗的目的。Mo 基催化剂作为研究最为广泛的耐硫催化剂, 活性组分为Mo
Mo基耐硫甲烷合成催化剂, 主要活性组分为MoS2, 常用载体有Al2O3、ZrO2、镁铝尖晶石以及Al2O3/ZrO2复合氧化物等, 添加Co、Ni、Ti、W、K等助剂。Whitehurst D D等[10]以Co(Ni)MoSx/Al2O3系列催化剂为研究对象, 综述几种主要结构模型的特点, 结果发现Co-Mo-S结构可能是某种真实活性相的前驱体, 而催化活性中心在实际催化反应过程中是动态的。
Daage M等[11]在MoS2催化剂结构与性能关系研究中提出Rim-Edge模型。该理论认为MoS2催化剂上存在两种类型的活性位, 即“ Rim” 和“ Edge” 活性位, “ Rim” 活性位位于MoS2晶片堆积的顶层和底层的边缘位置; “ Edge” 活性位位于晶片堆积层的中间夹层的侧面边缘部分。“ Rim” 活性中心具有较好的加氢活性。
协同作用模型[12]认为体相MoS2和助剂硫化物(如Co9S8)接触边界上发生的电子迁移为活性效应提供保障。整个体系处于动态平衡状态, 助剂硫化物实现对硫化钼活性中心数目及状态的调控。
嵌入模型[13]认为MoS2活性相与载体之间不存在相互作用, 但助剂和MoS2之间存在化学作用。在MoS2结构中, 硫原子上下六方紧密堆积, 阳离子处在六重三棱柱包围之中, 层间以范德华力结合, Co(Ni)离子占据范德华层的八面体位置, 助剂(Co和Ni)在体相MoS2的S-Mo-S层间边缘的八面体位置插入, 处于边缘位置, 由于缺硫而暴露的Mo3+是催化活性中心。Karroua M等[14]发现, 直接将Co9O8与MoS2均匀混合也表现出了良好的助催化效应, 这很难用嵌入模型解释。
该模型[15]认为焙烧过的氧化态催化剂中Mo物种与Al2O3载体通过Al2O3表面的-OH基团结合形成单分子层。Mo6+离子与单分子层上部“ 覆盖层” 中O2-离子结合抵消多余正电荷, 钴以Co2+形式取代与Mo单分子层相邻的表面Al3+离子, 占据Al2O3表面四面体位置, 提高Mo单分子层的稳定性。硫化后, S2-取代“ 覆盖层” 中的O2-。在临氢条件下, 移除部分S2-使相邻的Mo6+还原为Mo3+, Mo3+被认为是加氢脱硫反应的活性中心。
Co-Mo-S模型是接受度最广的结构模型。文献[16, 17, 18]借助谱穆斯堡尔谱和红外光谱技术等直接观察到Co-Mo催化剂中的Co-Mo-S相, 这一观点的提出, 对Co-Mo催化剂结构的研究具有重要意义, 证明催化剂与助剂之间的化学作用是存在的。Co-Mo-S结构是一种簇状结构[19], Co占据MoS2晶相的棱边位置, 三种元素的含量与化学计量比无太大关系。
Co-Mo-S结构存在两种不同类型[20], 即Co-Mo-S Ⅰ 型和 Co-Mo-S Ⅱ 型。以Al2O3担载的Mo/Co催化剂为例, 在Co-Mo-S Ⅰ 型中, Mo以单层晶片结构存在, 通过Mo-O-Al键与载体有强的相互作用, 催化剂硫化实验中发现, 这种结构不易被完全硫化; 在Co-Mo-S Ⅱ 型中, Mo以多层晶片结构存在, 与载体之间的作用力相对较弱, 易被完全硫化。由于空间效应的存在, Co-Mo-S Ⅰ 型中催化剂与载体成键有可能阻碍反应物分子在催化剂活性位上的吸附速率, 因此Co-Mo-S Ⅱ 型结构催化剂活性比Co-Mo-S Ⅰ 型结构催化更高。有学者发现Co-Mo-S模型存在缺陷[21], Co-Mo-S相只存在于新鲜催化剂中, 在加氢反应过程中极不稳定, 表明Co-Mo-S相很可能是某种真实活性相的前驱体。
Startsev A N等[22]提出了硫化的双金属模型。该模型中提到的Co: Mo物质的量比为1: 2, 在双金属模型中, 由于各物种间存在相互作用, 比单独的金属硫化物具有更好的催化活性。Chianelli R R等[23]认为, 在金属硫化物结合之后会产生某种特殊的电子效应, 这种电子效应能够平衡金属元素与S元素间以及金属元素与-SH2基团之间的键能大小, 这种动态平衡机制为加氢脱硫反应提供更多的催化活性位。
研究者对Mo基催化剂活性相的结构模型做了大量工作, 提出了多种结构模型, 虽然各结构模型均具有一定的局限性, 但这些模型都认为反应活性与催化剂表面的配位不饱和位中心的数量和分布密切相关[24], 并且越来越多的研究学者认为催化活性中心在实际催化反应中不再是静态的而是动态的。
Ni基催化剂表面甲烷合成反应的机理研究已有很多, 但Mo基催化剂表面耐硫甲烷合成反应的机理研究较少。
范崇正等[25]设计了一套脉冲实验装置考察MoS2催化甲烷化反应。实验结果表明该反应很可能是通过表面活化配合物进行的, 即CO与H2吸附在催化剂表面形成M-CHOH 烯醇式结构, 再与H2反应生成甲烷。
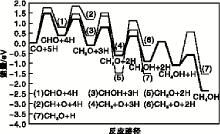
以朗格缪尔-欣谢尔伍德机理(L-H机理)为基础, Shi X R等[26]采用双S-Mo-S 薄层模型对Al2O3负载MoS2催化剂(10-10)晶面上的CO 加氢反应进行DFT 计算, 确定了最佳反应路径, 如图1所示。H2可在MoS2表面快速完成化学吸附, CO首先在MoS2表面边缘的Mo活性位上吸附, 与S活性位上吸附的H形成第一中间产物CHO。在相邻Mo活性位上以桥联形式吸附的CHO不发生CH与O解离, 继续与S活性位上吸附的H作用生产CH2O。CH2O作为第二种中间产物继续与H作用形成两种可能的中间产物CH3O和CH2OH。虽然CH3O和CH2OH在热力学上均是可行的, 但经计算生成CH2OH的过程在动力学上更有利。CH2OH发生C— O键的断裂, 生成CH2和OH, CH2物种中的C与相邻的两个Mo活性位以桥联方式连接, 与相离最近的S上活化的H继续反应生成CH3, 进一步反应生成CH4后在催化剂表面完成脱附。
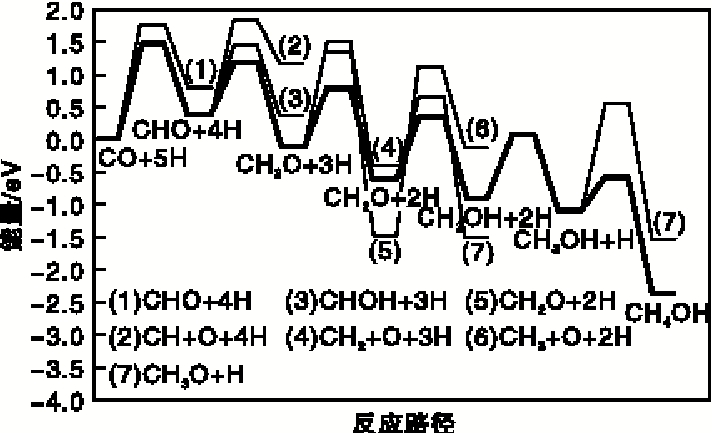 | 图1 MoS2表面CO加氢反应路径Figure 1 Reaction path of CO hydrogenation on surface of MoS2 |
Shi X R等[27]报道纯MoS2表面水汽变换的反应历程, 第一步为H2O物种分解成OH和H; 第二步为OH物种分解成O和H, 而不是CO与OH反应形成甲酸HCOO物种; 第三步为O物种与表面CO反应转化为亚稳态的CO2; 第四步为生成的CO2从催化剂表面脱附过程。
And H T等[28]认为β -Mo2C (001) 表面上水汽变换反应的速率控制步骤为CO与O转化为CO2的反应。HCOO物种最有可能的形成途径是CO2与H反应生成, HCOO进一步与表面H作用生成H2COO, 而非断裂其C— O键生成CHO和O物种, 生成的H2COO破坏HCOO的C— O键生成CH2O和O物种。
上述反应历程中出现的CH2O物种与CO甲烷化反应历程中的物种一致, 表明在耐硫甲烷化反应体系中, 同时存在CO、H2O和H2的情况下, 即CO甲烷化反应与水汽变换反应同时进行时, 两个反应过程是耦合在一起的, 需要考虑两种反应间的关联性。伏义路等[29]研究Ni基催化剂上变换-甲烷化反应体系。结果表明, 变换-甲烷化反应机理与甲烷化反应或变换反应有所不同, 把变换-甲烷化反应看成是变换反应和甲烷化反应的简单加合是不全面的。因此, Mo基催化剂表面变换-甲烷化反应的机理需要进一步深入研究。
水汽变换反应中, 反应气中的H2S是维持酸性变换催化剂活性的重要成分[30, 31]。在H2S气氛下, Sasaki T等[32]研究了Ni/Mo/Ti催化剂上水汽变换的反应过程, 可能进行的反应有:
MoS2 +H2O→ MoSO+H2S (路径1)
MoSO+CO→ MoS+CO2 (路径2)
MoS+H2O→ MoSO+H2 (路径3)
MoS+H2S→ MoS2 +H2 (路径4)
实验验证水汽变换反应是按照1、2、3路径进行的, 表明原料气中存在的H2S组分对Mo基催化剂上的耐硫甲烷化反应体系有利。
(1) Mo基催化剂的结构模型均存在一定的局限性, 包括最被认可的“ Co-Mo-S模型” , 提出更加合理的Mo基催化剂结构模型是一项重大挑战。
(2) Mo基催化剂表面CO甲烷化反应机理的研究需要针对不同的催化体系进行深入探讨, 包括多组分共同作用的Mo基催化作用机理以及Mo基催化剂表面积炭失活机理等方面的研究。
(3) 分析Mo基催化剂表面水汽变换反应历程发现生成的中间物种与甲烷化反应历程中的某些物种存在关联性, 需要进一步深入研究。原料气中H2S的存在可能促进耐硫甲烷化反应的进行。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|