
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
氢等离子体法制备双金属Ni-MoP催化剂
引用本文
李平. 氢等离子体法制备双金属Ni-MoP催化剂[J]. 工业催化, 2019,27(4): 76-79.
Li Ping. Preparation of bimetallic Ni-MoP catalyst by hydrogen plasma[J]. Industrial Catalysis, 2019,27(4): 76-79.
DOI:10.3969/j.issn.1008-1143.2019.04.014
Li Ping. Preparation of bimetallic Ni-MoP catalyst by hydrogen plasma[J]. Industrial Catalysis, 2019,27(4): 76-79.
Permissions
Copyright©2019, 《工业催化》编辑部
《工业催化》编辑部 所有
氢等离子体法制备双金属Ni-MoP催化剂
作者简介:李 平,1983年生,男,宁夏回族自治区银川市人,硕士, 讲师,研究方向为功能材料及燃料油精制。
摘要
以磷酸氢二胺和钼酸铵为原料,经氢等离子体法还原制备出单金属MoP及双金属Ni-MoP催化剂。采用XRD和SEM对催化剂进行分析表征;并在固定床反应器内以模拟油为研究对象评价催化剂活性。结果表明,双金属Ni-MoP催化剂的活性高于单金属MoP催化剂, 二苯并噻吩转化率达到26.21%,联苯选择性达35.61%,环己烷基苯选择性达19.96%。
关键词:
催化化学; 氢等离子; 磷化钼; Ni-MoP催化剂; 加氢脱硫
中图分类号:TE624.9;TQ426.95
文献标志码:A
文章编号:1008-1143(2019)04-0076-04
Preparation of bimetallic Ni-MoP catalyst by hydrogen plasma
Abstract
MoP and Ni-MoP catalysts were prepared by hydrogen plasma reduction using hydrogen diamine phosphate and ammonium molybdate as raw material.The catalyst was characterized by XRD and SEM,and evaluated in a fixed-bed reactor with simulated oil used as research object.The results showed that the activity of modified Ni-MoP catalyst was higher than MoP catalyst.The conversion of dibenzothiophene was 26.21%,selectivity of biphenyl and cyclohexalkylbenzene were 35.61% and 19.96% reapectively.
Keyword:
catalysis chemistry; hydrogen plasma; molybdenum phosphide; hydrodesulfurization
随着人们环保意识的增强, 对燃料油中硫含量提出了严苛的要求, 因此燃料油脱硫技术成为研究热点[1]。磷化物催化剂在燃料油催化加氢脱硫方面具有诸多优点, 研究者不断尝试磷化物催化剂的制备及改性[2, 3]。等离子体由于具有高能粒子, 引起了广泛关注, 并将其应用到催化剂制备中[4]。本文以磷酸氢二胺和钼酸铵为原料, 经氢等离子体法还原制备单金属MoP及双金属Ni-MoP催化剂。采用XRD和SEM对催化剂进行表征; 并在固定床反应器内以模拟油为研究对象评价催化剂催化活性。
1 实验部分
1.1 试剂与仪器
硝酸镍、钼酸铵、磷酸氢二铵和硝酸均为分析纯; 氢气, 99.99%。
自制等离子还原反应装置, 马弗炉SX2-2.5-10, 粉末压片机769YP-24B。
1.2 催化剂制备
1.2.1 前驱体的制备
称取一定量的磷酸氢二铵, 钼酸铵(改性试剂硝酸镍, 0.05%)依次加入去离子水中, 在温水浴中完全溶解、搅拌、蒸干, 在120 ℃下干燥12 h, 500 ℃下焙烧4 h, 自然冷却至室温, 制得催化剂氧化物前驱体。
1.2.2 氢等离子还原
将一定量的氧化物前驱体装入介质阻挡放电等离子体反应器内, 限定放电频率约10 kHz, 调节等离子体发生器的输入电压及输入功率, 设定H2流速为130 mL· min-1。按每5 V的频率持续递增升压, 到达最佳电压时反应2 h。
1.3 催化剂表征
采用日本理学公司Dmax200PC型X衍射光谱仪测定各样品晶体结构; 采用XL-30型电子显微镜观察催化剂表面形态。
1.4 实验装置
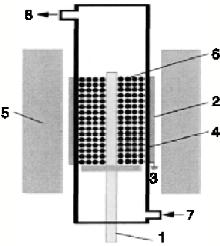
催化剂前驱体还原反应在质阻挡放电反应器内进行, 放电产生氢非平衡等离子体, 用于还原氧化物前驱体, 反应器结构如图1所示。
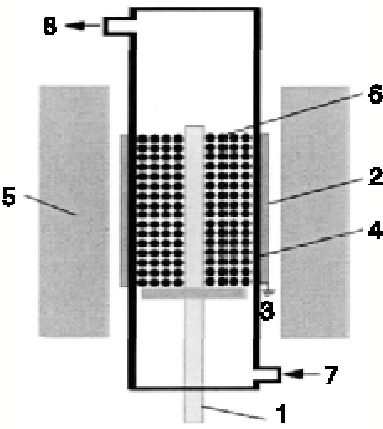 | 图1 介质阻挡放电反应器结构 1.高压电极; 2.接地极; 3.地面; 4.石英管; 5.绝热层; 6.催化剂前体床层; 7.气体入口; 8.气体出口Figure 1 Structure of dielectric barrier discharge reactor |
以燃料油中最难脱出的二苯并噻吩(DBT)为考察对象, 配制二苯并噻吩质量分数0.8%的十氢萘溶液为模拟油。加氢脱硫反应在内径为8 mm的固定床反应器内进行。采用气相色谱仪分析产物组成, 计算二苯并噻吩转化率和产物选择性。
2 结果与讨论
2.1 XRD
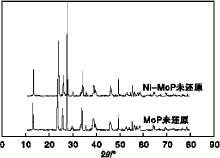
图2为MoP与Ni-MoP催化剂氧化物前驱体的XRD图。由图2可以看出, 未还原的MoP催化剂与Ni-MoP催化剂的特征峰基本一致, 均出现了MoO3的特征峰, 说明Ni均匀分散到磷化物催化剂内。
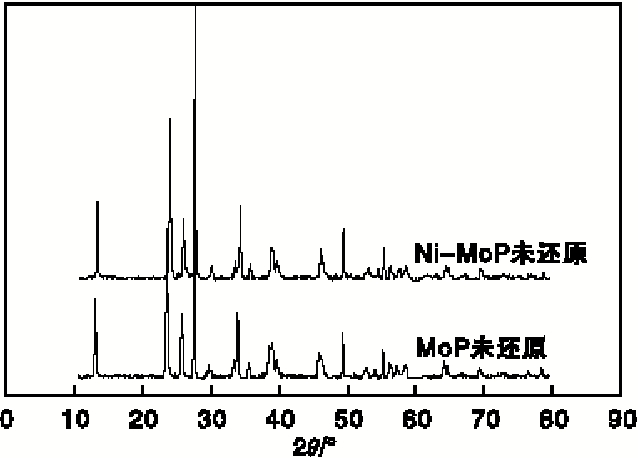 | 图2 MoP与Ni-MoP催化剂氧化物前驱体的XRD图Figure 2 XRD diagrams of MoP and oxide precursor of Ni-MoP catalyst |
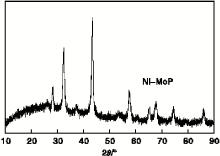
图3为Ni-MoP催化剂的XRD图。由图3可以看出, 在2θ =28° 、32° 、43° 、57° 、65° 、67° 和74° 等出现了与MoP标准图谱一致的特征峰。说明经过氢等离子体法可以还原出Ni-MoP催化剂。
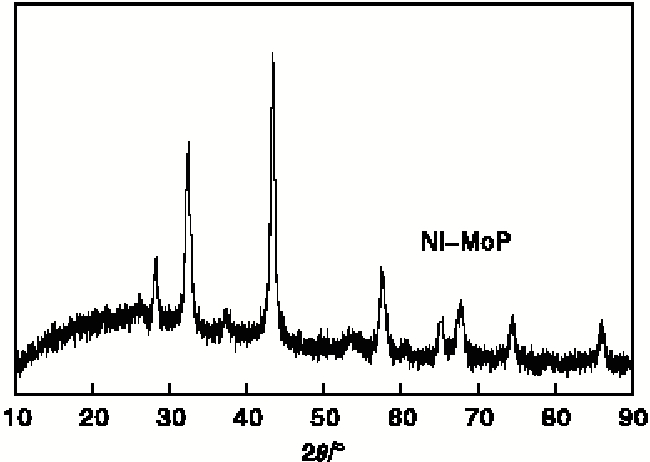 | 图3 Ni-MoP催化剂的XRD图Figure 3 XRD diagram of Ni-MoP catalyst |
2.2 SEM
图4为不同放大倍数Ni-MoP催化剂的SEM照片。从图4可见, 放大5000倍的电镜照片, 催化剂呈小颗粒状, 其粒径在(1~3) μ m, 大部分颗粒粒径分布均匀; 放大10000倍的电镜照片, 其表面呈凹凸状、颗粒之间有孔道, 这种表面结构有助于增大比表面积, 提高催化剂活性。
 | 图4 不同放大倍数Ni-MoP催化剂的SEM照片Figure 4 SEM images of Ni-MoP catalysts at different magnification |
2.3 还原电压
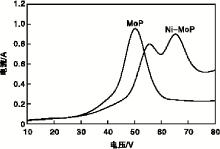
MoP与Ni-MoP催化剂还原电压与电流关系见图5。由图5可知, 当电压低于40 V时, 能量较小无法使反应器内氢气等离子化, 故产生微弱的电流; 继续升高电压, 电流急剧增加, 在(50~65) V时电流最大, 说明已经达到氢气的击穿电压, 反应器内呈现氢等离子体状态[5]。但由于催化剂MoP和Ni-MoP氧化物前驱体组成不同, 致使Ni-MoP条件下的高电流(> 0.7A)区间高于MoP高电流区间, 也说明 Ni-MoP条件下产生的氢等离子体密度更高, 这有助于Ni-MoP催化剂氧化物前驱体的还原, 从而提高催化剂活性。
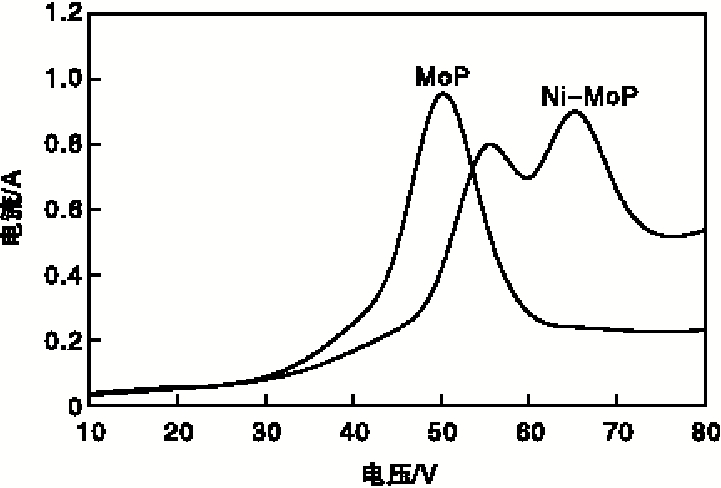 | 图5 MoP和Ni-MoP催化剂还原电压与电流关系Figure 5 Relationship between voltage and current of MoP and Ni-MoP catalyst |
2.4 催化剂催化活性
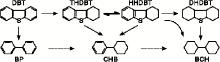
一般认为, 二苯并噻吩的加氢脱硫按照两个平行的反应路径进行[6], 如图6所示。
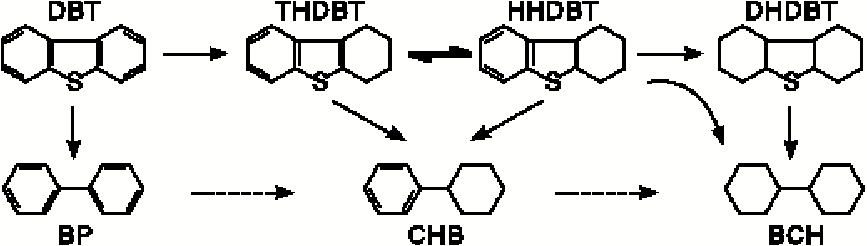 | 图6 二苯并噻吩的加氢脱硫反应网路Figure 6 Hydrodesulfurization reaction network of dibenzothiophene |
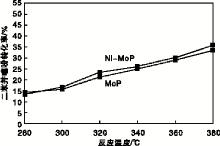
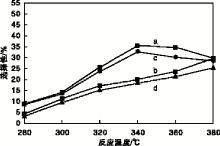
实验以二苯并噻吩转化率、直接脱硫路径产物联苯(BP)选择性及预加氢反应路径产物环己烷基苯(CHB)选择性为指标, 评价催化剂加氢脱硫活性, 结果如图7和图8所示。从图7和图8可以看出, 随着反应温度的升高, 两种催化剂上二苯并噻吩转化率均随之升高, 联苯选择性在340 ℃达到最大后逐渐减小; 环己烷基苯选择性处于增加趋势, 但Ni-MoP催化剂上的二苯并噻吩转化率、联苯与环己烷基苯的选择性均高于MoP催化剂; 说明助剂Ni与Mo产生了协同效应[7], 促使活性组分MoP的生长, 从而提高了Ni-MoP催化剂的加氢脱硫活性。在340 ℃条件下, Ni-MoP催化剂可使二苯并噻吩转化率达到26.21%, 联苯选择性达35.61%, 环己烷基苯选择性达19.96%。
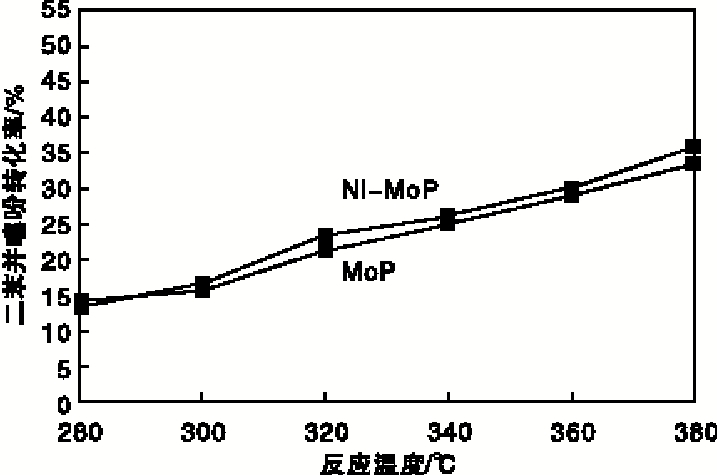 | 图7 反应温度对二苯并噻吩转化率的影响Figure 7 Effect of reaction temperature on conversion of dibenzothiophene |
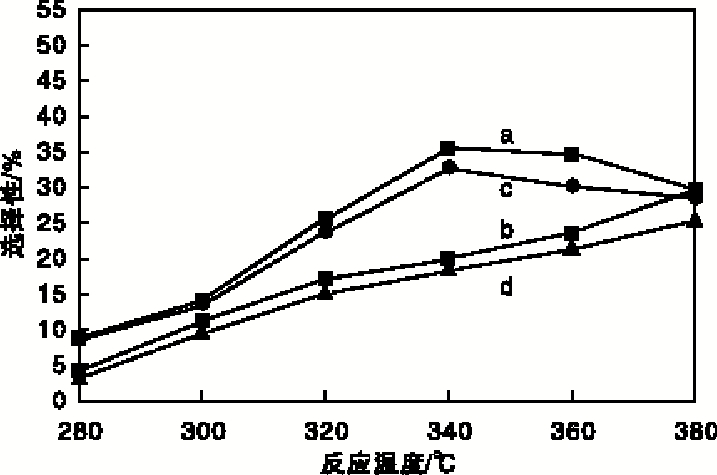 | 图8 反应温度对联苯和环己烷基苯选择性的影响 a.Ni-MoP催化剂上联苯选择性; b. Ni-MoP催化剂上环已烷基苯选择性; c.MoP催化剂上联苯选择性; d. MoP催化剂上环已烷基苯选择性Figure 8 Effect of reaction temperature on selectivity of biphenyl and cyclohexylbenzene |
3 结 论
采用氢等离子体技术均可制备出磷化物催化剂, 添加助剂Ni改性后的Ni-MoP催化剂活性高于未改性MoP催化剂。在340 ℃条件下, Ni-MoP催化剂可使二苯并噻吩的转化率达到26.21%, 联苯选择性达35.61%, 环己烷基苯选择性达19.96%。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|