




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
单斜相与四方相混合晶相组成ZrO2负载镍催化剂催化顺酐选择性加氢
引用本文
赵丽丽, 张因, 吴天杰, 赵敏, 赵江红, 王永钊, 赵永祥. 单斜相与四方相混合晶相组成ZrO2负载镍催化剂催化顺酐选择性加氢 [J]. 工业催化, 2019,27(5): 31-38.
Zhao Lili, Zhang Yin, Wu Tianjie, Zhao Min, Zhao Jianghong, Wang Yongzhao, Zhao Yongxiang. Selective hydrogenation of maleic anhydride catalyzed by supported nickel catalysts on ZrO2 of monoclinic and tetragonal mixed crystal phase [J]. Industrial Catalysis, 2019,27(5): 31-38.
DOI:10.3969/j.issn.1008-1143.2019.05.007
Zhao Lili, Zhang Yin, Wu Tianjie, Zhao Min, Zhao Jianghong, Wang Yongzhao, Zhao Yongxiang. Selective hydrogenation of maleic anhydride catalyzed by supported nickel catalysts on ZrO2 of monoclinic and tetragonal mixed crystal phase [J]. Industrial Catalysis, 2019,27(5): 31-38.
Permissions
Copyright©2019, 《工业催化》编辑部
《工业催化》编辑部 所有
单斜相与四方相混合晶相组成ZrO2负载镍催化剂催化顺酐选择性加氢
作者简介:赵丽丽,1982年生,女,山东省乐陵市人,在读博士研究生,助教,研究方向为多相催化。
摘要
通过调控水热法制备条件制备同为单斜相和四方相混合晶相组成、但织构性质和表面结构性质不同的两种ZrO2载体,采用浸渍法制备镍质量分数为10%的Ni/ZrO2催化剂,考察不同反应温度[(150~240) ℃]和氢气压力[(3~7) MPa] 条件下两种ZrO2载体负载镍催化剂的顺酐加氢性能。采用XRD、H2-TPR、H2-TPD和拉曼光谱等对催化剂进行表征。结果表明,与镍物种发生较强相互作用的ZrO2负载镍催化剂具有较高的C=C键加氢活性与选择性,几乎没有C=O加氢活性,在所考察的反应温度和反应压力范围,催化剂上丁二酸酐选择性均高于95.1%,γ-丁内酯选择性均低于4.9%。与之不同,与镍物种发生较弱相互作用的ZrO2负载镍催化剂具有较弱的C=C键加氢活性,然而,该催化剂表现出一定的C=O加氢活性,并且其C=O加氢活性随反应温度或反应压力的提高而显著提高。在反应温度240 ℃、氢气压力5 MPa条件下,γ-丁内酯选择性高达60.6%。推测晶相组成相似的两种ZrO2载体负载镍催化剂明显的C=O加氢性能差异与其表面结构性质不同有关。
关键词:
催化化学; Ni/ZrO2催化剂; 顺酐; 选择性加氢; 表面结构; 氧空位
中图分类号:O643.36;TQ426.6
文献标志码:A
文章编号:1008-1143(2019)05-0031-08
Selective hydrogenation of maleic anhydride catalyzed by supported nickel catalysts on ZrO2 of monoclinic and tetragonal mixed crystal phase
Abstract
Two kinds of ZrO2 supports,both composed of monoclinic and tetragonal phases,but with different texture properties and surface structures,were prepared by adjusting hydrothermal conditions.Ni/ZrO2 catalysts mith Ni mass fraction of 10% were prepared using an impregnation method and were characterized by XRD,H2-TPR,H2-TPD and Raman techniques.Their catalytic performances in maleic anhydride hydrogenation were tested under different reaction temperature (150-240) ℃ and H2 pressure (3-7) MPa.Results showed that Ni/ZrO2 catalyst with strong interaction of nickel species and ZrO2 support had higher C=C hydrogenation activity,whereas it had almost no C=O hydrogenation activity.Within the range of reaction temperature and H2 pressure investigated,the selectivity of the catalyst for succinic anhydride was above 95.1%,correspondingly,the selectivity of γ-butyrolactone was less than 4.9%.Ni/ZrO2 catalyst with weaker interaction of nickel species and ZrO2 support had lower C=C hydrogenation activity,whereas it exhibited C=O hydrogenation,and the C=O hydrogenation activity increased significantly with rise of reaction temperature and H2 pressure.The selectivity towards γ-butyrolacetone was as high as 60.6% at reaction temperature of 210 ℃ and H2 pressure of 5 MPa.It was inferred that the obvious difference of C=O hydrogenation activity of two ZrO2 supported nickel catalysts was related to the different surface structure properties of ZrO2 supports.
Keyword:
catalytic chemistry; Ni/ZrO2 catalysts; maleic anhydride; selective hydrogenation; surface structure; oxygen vacancies
顺酐作为三大酸酐之一, 可被加氢获得丁二酸酐、γ -丁内酯、1, 4-丁二醇和四氢呋喃等一系列有重要用途的高附加值化学品[1]。其中, 丁二酸酐是生产新型可降解塑料聚丁二酸丁二醇酯的主要原料, 而γ -丁内酯作为低毒环保型溶剂被广泛应用[2]。
顺酐是典型的α , β -不饱和羰基化合物, 分子中含有C=C、C=O、C— O— C官能团, 呈五元环状结构, 共轭效应导致电子云密度趋于均一, 因而仅对顺酐分子中C=C双键加氢合成丁二酸酐或同时对C=C双键和一个C=O键加氢合成γ -丁内酯比较困难[3]。因此, 设计与制备高效选择性加氢催化剂, 实现不同加氢产物丁二酸酐/γ -丁内酯的高选择性合成, 探究催化剂结构与性能之间的关系规律, 具有重要的科学意义与实际价值。
通常应用于顺酐加氢反应的催化剂主要包括贵金属Pd、Pt、Ru基催化剂和非贵金属Ni、Cu基催化剂[4, 5, 6, 7], Ni基催化剂因其较高的加氢活性及低的成本而被广泛应用于顺酐加氢反应中, 所用Ni基催化剂载体以Al2O3、SiO2、SiO2-Al2O3、黏土、TiO2和CeO2等居多[8, 9, 10, 11], 其中可还原性氧化物TiO2、CeO2负载Ni基催化剂在顺酐加氢反应中表现出优异的选择性加氢性能, 推测催化剂还原过程中产生的氧空位促进了C=O加氢[12, 13]。本课题组制备了同为单斜相和四方相混合晶相组成的两种ZrO2载体[14], 并考察其负载Ni催化剂在反应温度210 ℃、氢气压力5 MPa条件下的顺酐加氢性能, 结果发现, 虽然两种ZrO2载体晶相结构组成相似, 但其负载Ni催化剂的顺酐加氢性能存在较大差异。
为进一步了解影响ZrO2载体负载镍催化剂顺酐加氢性能差异的原因、深入认识ZrO2载体及Ni/ZrO2催化剂表面结构对催化剂C=O加氢性能的影响, 本文考察反应条件对同为单斜相和四方相混合晶相组成的两种ZrO2载体制备的催化剂顺酐加氢性能的影响, 并结合XRD、H2-TPR、H2-TPD、拉曼光谱等探究载体及催化剂表面结构与其选择性加氢性能之间的内在关系。
1 实验部分
1.1 催化剂制备
1.1.1 ZrO2载体制备
ZrO2 (P)制备:100 ℃条件下回流0.5 mol· L-1的硝酸氧锆溶液240 h, 期间通过滴定氨水溶液保持溶液pH=1.5。然后, 将得到的含有白色沉淀物的溶液(80 mL)转移至100 mL水热釜中, 110 ℃晶化4 h。最终得到的沉淀物使用无水乙醇洗涤数次至溶液pH=7。将水洗后的沉淀物110 ℃干燥12 h, 空气气氛焙烧4 h, 制备的ZrO2标记为ZrO2 (P)。
ZrO2 (H)制备:取80 mL溶液于100 mL的水热釜中140 ℃晶化2.5 h, 降至室温, 使用无水乙醇洗涤沉淀物数次至溶液pH=7, 沉淀物于110 ℃干燥12 h, 空气气氛400 ℃焙烧4 h, 制备的ZrO2标记为ZrO2 (H)。
1.1.2 Ni/ZrO2催化剂制备
取0.5476 g硝酸镍和2.2 mL蒸馏水配置成硝酸镍溶液, 向硝酸镍溶液中加入1 g的ZrO2 (P) 或ZrO2 (H)载体, 超声30 min, 静置3 h, 于110 ℃干燥12 h, 空气气氛450 ℃焙烧4 h, 得到氧化态催化剂分别标记为NiO/ZrO2 (P)和NiO/ZrO2 (H)。氧化态催化剂在H2气氛中400 ℃还原3 h, 制得Ni质量分数为10%的ZrO2载体负载镍催化剂, 分别标记为Ni/ZrO2 (P)和Ni/ZrO2 (H)。
1.2 顺酐加氢性能评价
Ni/ZrO2催化剂的顺酐加氢性能评价测试在大连通产仪器仪表有限公司FXY-0.1型间歇式高压釜中进行。测试前, 首先将氧化态催化剂在H2气氛中400 ℃还原3 h, 待降至室温, 在N2吹扫保护下将还原后的催化剂转移至四氢呋喃溶液中(40 mL 四氢呋喃+4.9 g 顺酐)。将包含有催化剂的四氢呋喃溶液转移至100 mL高压釜中, 密封釜体, 分别使用N2和H2分别置换釜内空气和N2, 保证釜内为纯H2气氛, 打开反应釜搅拌装置(400 r· min-1), 开始计时反应。反应初期1 h内每20 min取样一次, 反应1 h后, 每1 h取样一次。
反应产物在安捷伦7890B气相色谱仪(DB-5色谱柱, FID检测器)上分析测试, 计算顺酐转化率和产物选择性。
1.3 催化剂表征
XRD测试在德国布鲁克公司 D8 Advance 型X射线衍射仪上进行, Cu靶, 工作电压为为40 kV, 工作电流40 mA, 扫描范围10° ~80° , 扫描速率为3 ° · min-1。
低温氮气物理吸附-脱附表征采用美国麦克仪器公司ASAP-2020物理吸附仪, 样品在250 ℃高真空处理3 h, 然后置于液氮中冷却至-196 ℃进行测试, BET方程计算样品的比表面积。
在美国麦克仪器公司AutoChem2920化学吸附仪上进行H2-TPR测试, 取30 mg样品于纯Ar气氛中300 ℃处理1 h, 然后降至50 ℃, 切换至10%H2/Ar混合气 (30 mL· min-1)气氛, 以升温速率10 ℃· min-1升温至700 ℃, TCD检测器记录信号。
H2-TPD采用美国麦克仪器公司AutoChem2920化学吸附仪, 取102.7 mg样品在纯H2气氛(30 mL· min-1)中400 ℃还原3 h(升温速率为10 ℃· min-1), 然后切换至纯Ar气氛50 ℃吹扫1 h。待样品温度降至50 ℃, 切换至10%H2-Ar混合气 (30 mL· min-1)气氛使其吸附H2至饱和。之后切换至纯Ar气氛直至基线稳定。然后以10 ℃· min-1的升温速率升温至700 ℃, 同时记录脱出物响应信号。
拉曼光谱测试采用Horiba LabRam HR Evoluton 紫外共焦拉曼光谱仪, 激光波长532 nm, 扫描范围(100~1000) cm-1。根据以下公式计算ZrO2表面结构中四方相含量[15]:
X(T) =
式中, I(T)为拉曼光谱中147 cm-1和269 cm-1处拉曼峰峰强度之和; I(M)为拉曼光谱中176 cm-1和187 cm-1处拉曼峰峰强度之和。
2 结果与讨论
2.1 反应温度
在氢气压力5 MPa, 不同反应温度条件下Ni/ZrO2 (P)和Ni/ZrO2 (H)催化剂顺酐加氢性能随反应时间的变化规律如图1所示。
 | 图1 反应温度对Ni/ZrO2(P)和Ni/ZrO2(H)催化剂顺酐加氢性能的影响Figure 1 Effects of reaction temperature on catalytic performance of Ni/ZrO2(P) and Ni/ZrO2(H) catalysts in hydrogenation of maleic anhydride |
由图1可以看出, 在反应温度150 ℃、5 MPa氢气压力条件下, Ni/ZrO2 (P)催化剂上顺酐转化率高于Ni/ZrO2 (H)催化剂。反应时间20 min时, Ni/ZrO2 (P)催化剂上顺酐转化率为28.8%, 而Ni/ZrO2 (H)催化剂上顺酐转化率仅为18.3%。提高反应温度至210 ℃和240 ℃时, 两种催化剂上顺酐转化率均随反应温度的提高逐渐增大, 而且同一反应温度条件下, Ni/ZrO2 (P)催化剂上顺酐转化率总是高于Ni/ZrO2 (H)催化剂。在顺酐完全转化前, 两种催化剂的加氢产物均是丁二酸酐, 选择性高于98%, 表明顺酐首先发生C=C键加氢获得丁二酸酐, 即催化剂上顺酐转化率高低反映了催化剂C=C键加氢活性高低, 表明Ni/ZrO2 (P)催化剂的C=C键加氢活性显著高于Ni/ZrO2 (H)催化剂。
随反应时间延长, Ni/ZrO2 (P)和Ni/ZrO2 (H)催化剂呈现出不同的加氢产物分布。由图1还可以看到, 在考察的反应温度范围, Ni/ZrO2 (P)催化剂表现出高丁二酸酐选择性。当反应温度为150 ℃、反应时间240 min时, Ni/ZrO2(P)催化剂上丁二酸酐选择性为100%, 即使提高反应温度至240 ℃, 反应时间延长至480 min时, 丁二酸酐选择性仍高达95.1%, γ -丁内酯选择性低于4.9%, 表明Ni/ZrO2 (P)催化剂几乎没有C=O加氢活性。与此不同, Ni/ZrO2 (H)催化剂上丁二酸酐选择性随着反应温度的提高逐渐降低, γ -丁内酯选择性相应逐渐升高。反应温度150 ℃、反应时间480 h时, γ -丁内酯选择性为20.1%, 当反应温度提高至240 ℃时, γ -丁内酯选择性增大至60.6%, 表明其C=O加氢活性随反应温度升高而逐渐增高。两种催化剂随反应温度变化呈现不同的加氢产物分布变化规律表明, Ni/ZrO2 (P)催化剂几乎没有C=O加氢活性, 而Ni/ZrO2 (H)催化剂有C=O加氢活性, 并且其C=O加氢活性随反应温度的升高而显著提高。
2.2 氢气压力
在反应温度210 ℃, 不同氢气压力条件下, Ni/ZrO2(P)和Ni/ZrO2(H)催化剂顺酐加氢性能如图2所示。由图2可以看到, 在考察的氢气压力范围, Ni/ZrO2 (P)催化剂上顺酐转化率明显高于Ni/ZrO2(H)催化剂, 表明Ni/ZrO2 (P)催化剂的C=C键加氢活性显著高于Ni/ZrO2 (H)催化剂。当氢气压力为3 MPa、反应时间为480 min时, Ni/ZrO2 (P)催化剂上丁二酸酐选择性为98.8%, 即使提高氢气压力至7 MPa, 丁二酸酐选择性仍高达96.8%, γ -丁内酯选择性低于4.9%。
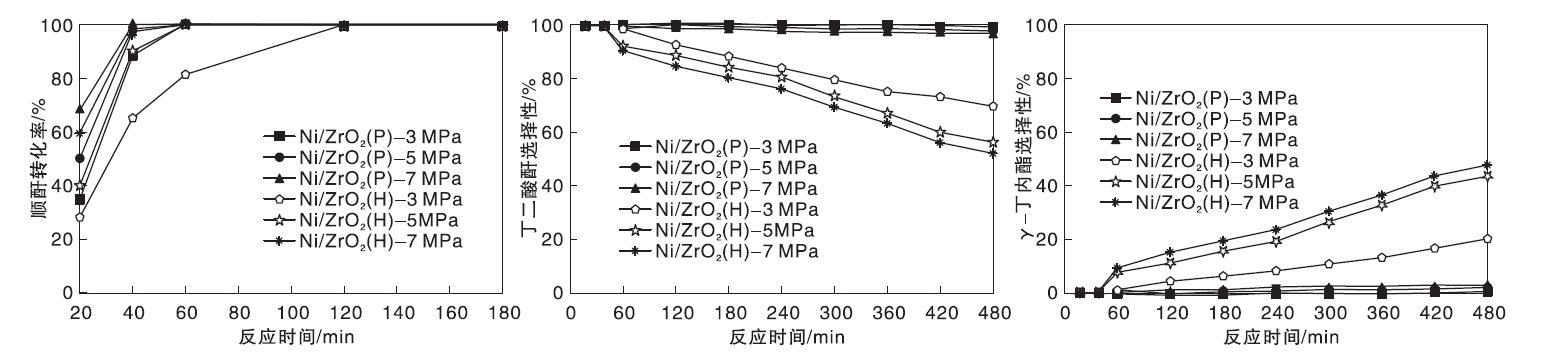 | 图2 H2压力对Ni/ZrO2(P)和Ni/ZrO2(H)催化剂顺酐加氢性能的影响Figure 2 Effects of H2 pressure on catalytic performance of Ni/ZrO2(P) and Ni/ZrO2(H) catalysts in hydrogenation of maleic anhydride |
与此变化规律不同, 当氢气压力为3 MPa时, Ni/ZrO2(H)催化剂上γ -丁内酯选择性为20.1%, 升高氢气压力至7 MPa时, γ -丁内酯选择性增至48.2%。关于氢气压力对两种催化剂顺酐加氢性能的影响再次证明, Ni/ZrO2 (P)催化剂几乎没有C=O加氢活性, 而Ni/ZrO2 (H)催化剂的C=O加氢活性随氢气压力提高而逐渐提高, 与不同反应温度条件下两种催化剂顺酐加氢性能考察结果一致。
分析结果发现, 对于Ni/ZrO2催化体系, 其顺酐加氢产物为丁二酸酐和γ -丁内酯, 未检测到其他深度加氢产物或副产物生成。
2.3 XRD
图3为ZrO2 (P)与ZrO2 (H)载体及其负载Ni催化剂的XRD图。由图3可以看到, ZrO2 (P)与ZrO2 (H)载体晶相结构均为单斜相和四方相的混合相, 且其单斜相特征峰峰强度与四方相特征峰峰强度之比接近, 表明两种载体晶相结构组成基本一致。在氧化态催化剂XRD图中, 出现NiO的特征衍射峰, 载体ZrO2的晶相结构未发生明显变化。催化剂经还原后, NiO特征衍射峰消失, 出现金属Ni的特征衍射峰。根据谢乐公式计算得氧化态催化剂和还原态催化剂中对应的NiO和Ni晶粒尺寸(见表1)。由表1可以看到, ZrO2(H)负载镍催化剂的NiO和Ni晶粒尺寸分别为26.1 nm、40.0 nm, 明显大于ZrO2(P)负载镍催化剂(分别为16.2 nm、18.2 nm), 这应该与ZrO2(P)载体较高的比表面积有关。
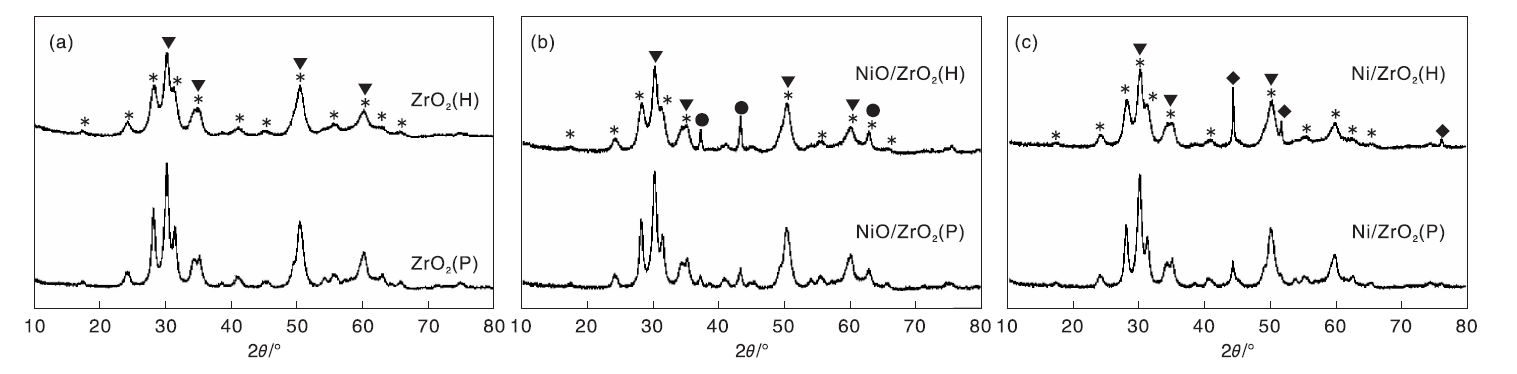 | 图3 ZrO2(P)和ZrO2(H)载体(a)、负载Ni氧化态催化剂(b)及还原后催化剂(c)的XRD图Figure 3 XRD patterns of ZrO2 (P) and ZrO2 (H) supports (a), calcined Ni catalysts(b) and reduced catalysts(c) |
| 表1 ZrO2 (P)与ZrO2 (H)载体及其负载Ni催化剂的表面织构性质 Table 1 Texture property of ZrO2 (P) and ZrO2 (H) supports and corresponding supported nickel catalysts |
2.4 H2-TPR
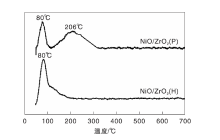
图4为ZrO2(P)和ZrO2(H)载体及其负载Ni催化剂的H2-TPR谱图。由图4可以看出, ZrO2 (H)载体在(100~700) ℃的检测范围无耗氢峰出现, ZrO2 (P)载体在540 ℃处出现峰强度较弱、宽泛的耗氢峰, 表明ZrO2 (P)载体中的氧物种较ZrO2 (H)载体更易还原。文献报道[16], ZrO2表面的低配位氧离子更易被还原, 而且ZrO2表面的缺陷结构-氧空位会促进这种氧物种的还原。由此推测, ZrO2(P)载体表面有较多的低配位氧离子和氧空位。
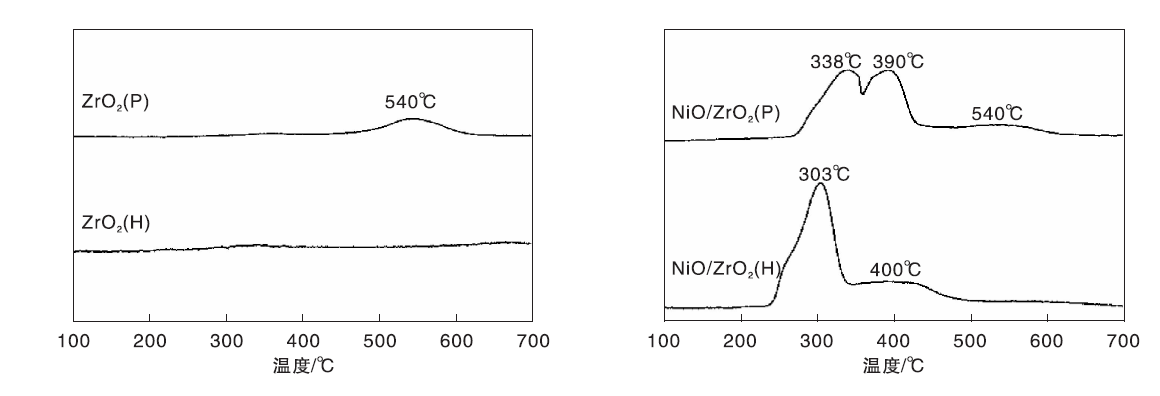 | 图4 ZrO2(P)和ZrO2(H)载体及其负载Ni催化剂的H2-TPR谱图Figure 4 H2-TPR profiles of ZrO2 (P) and ZrO2 (H) supports and corresponding supported nickel catalysts |
由图4还可以看出, NiO/ZrO2(H)催化剂在303 ℃处出现主耗氢峰, 同时在400 ℃处出现一弱的肩峰, 其中303 ℃处的耗氢峰归属为与体相NiO类似的镍物种的还原, 而400 ℃处的肩峰则归属为与载体ZrO2(H)有弱相互作用的镍物种的还原[17]。NiO/ZrO2(P)催化剂在338 ℃、390 ℃处呈现两个连续的耗氢峰, 同时在540 ℃处出现弱的耗氢峰, 其中, 338 ℃处的耗氢峰归属为与载体ZrO2(P)存在弱相互作用的镍物种的还原, 而390 ℃处的耗氢峰可归属为与载体ZrO2(P)发生较强相互作用的镍物种的还原[17], 540 ℃处的耗氢峰应是与其载体一致的耗氢峰。
与NiO/ZrO2(H)催化剂的还原峰相比, NiO/ZrO2(P)催化剂的还原峰均向高温方向位移, 且较高温度处的还原峰峰面积明显增大, 表明镍物种与ZrO2(P)载体的相互作用增强, 而且与ZrO2(P)载体发生较强相互作用的镍物种较多。Puigdollers A R等[16]的密度泛函理论计算结果表明, 富含低配位氧离子和氧空位的ZrO2与负载金属之间的相互作用更强。因此, ZrO2(P)载体表面有更多的低配位氧离子和氧空位, 与镍物种较易产生较强的金属-载体相互作用, 有利于镍物种锚定在载体表面, 抑制高温焙烧还原过程中镍晶粒的迁移聚集, 一定程度上促进Ni的分散。因而Ni/ZrO2(P)催化剂中较小的Ni晶粒尺寸应该也与镍物种和ZrO2(P)载体较强的相互作用有关。
2.5 拉曼光谱
与XRD相比, 拉曼光谱表征对样品的中程有序性及分子结构中氧的位移更灵敏, 因此拉曼表征常被用来分析ZrO2样品表面微观结构的变化[18]。图5为ZrO2 (P)与ZrO2 (H)载体及其负载Ni催化剂的拉曼光谱图。
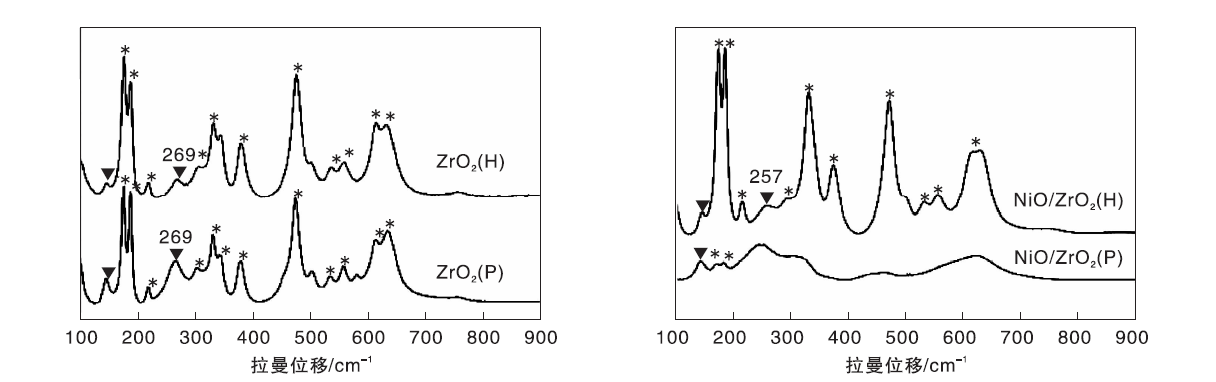 | 图5 ZrO2 (P)与ZrO2 (H)载体及其负载Ni催化剂的拉曼光谱图Figure 5 Raman spectra of ZrO2 (P) and ZrO2 (H) supports and corresponding supported nickel catalysts |
由图5可以看到, ZrO2(P)与ZrO2(H)载体表面结构均为四方相和单斜相的混合相[19], 但四方相(147 cm-1、269 cm-1)特征拉曼振动峰与单斜相特征拉曼振动峰(176 cm-1、187 cm-1)峰强度之比不同, 表明两种载体表面四方相含量不同。根据拉曼光谱中四方相含量计算公式计算知ZrO2 (P)载体表面四方相含量为23%, ZrO2 (H)载体表面四方相含量仅11%。基于纳米粒子效应[20]— — 纳米ZrO2粒子在室温下能够以四方晶相形式存在是由于额外产生氧空位的原因, 可推测ZrO2 (P)载体表面具有更丰富的氧空位, 与H2-TPR表征结果一致。
由图5还可以看出, ZrO2(P)载体负载Ni后, 其四方相拉曼特征峰消失, 同时出现一连续宽泛的拉曼峰, 归属为无定形结构拉曼峰[21]。Tosoni S等[22]的密度泛函理论计算结果表明, 富含氧空位的ZrO2表面对金属Ni团簇的吸附能(-4.28 eV)远低于化学计量比ZrO2表面对Ni团簇的吸附能(-2.31 eV), 而且在氧空位分布于ZrO2表层的情况下, Ni团簇进入氧空位是其最稳定的存在方式。结合Puigdollers A R等[16]的理论计算结果及本文H2-TPR表征结果, 推测ZrO2(P)载体表面较多的低配位氧离子和氧空位是导致镍物种与ZrO2(P)载体发生较强相互作用的原因。同时这种较强的相互作用使得少量镍物种进入ZrO2载体表面氧空位或ZrO2晶胞, 导致Ni/ZrO2(P)催化剂表面氧空位减少, 四方相结构无序化[14]。
与ZrO2(P)载体负载Ni后拉曼光谱图变化不同, ZrO2(H)载体负载Ni后, 其表面结构仍然是单斜相和四方相的混合相, 但可明显看到ZrO2(H)载体拉曼光谱图中269 cm-1拉曼峰位移至Ni/ZrO2(H)催化剂的257 cm-1处。269 cm-1处拉曼峰是四方相ZrO2的最强特征峰, 归属为非对称的Zr-O-Zr伸缩振动峰。文献报道[23], 该峰向低波数位移, 是由于氧空位的增加使得四方相结构对称性降低的结果。这表明镍物种与ZrO2(H)载体弱相互作用促进了Ni/ZrO2(H)催化剂表面新的氧空位的产生。
2.6 H2-TPD
图6为Ni/ZrO2(P)和Ni/ZrO2(H)催化剂的H2-TPD谱图。由图6可以看出, Ni/ZrO2(H)催化剂在80 ℃处呈现强的氢脱附峰, 归属为弱吸附在活性金属Ni表面的氢物种的脱附[24]。与之不同, Ni/ZrO2(P)催化剂分别在80 ℃处出现峰强度较强的氢脱附峰和206 ℃处峰强度较弱、峰形宽化的氢脱附峰。文献报道[25], H2-TPD谱图中低于300 ℃的氢脱附峰均为吸附在活性金属Ni表面的氢物种的脱附, 其中206 ℃处的氢脱附峰可归属为与金属Ni有较强吸附作用的氢物种的脱附。氢脱附量计算结果表明, 在(50~210) ℃, Ni/ZrO2(P)催化剂氢脱附量是Ni/ZrO2(H)催化剂的1.12倍, 表明Ni/ZrO2(P)催化剂表面有更多的吸附活化氢位点。结合XRD和H2-TPR表征结果不难理解, Ni/ZrO2(P)催化剂中Ni晶粒尺寸较小, 分散度较高, 表面暴露的吸附活化氢的活性位点较多。
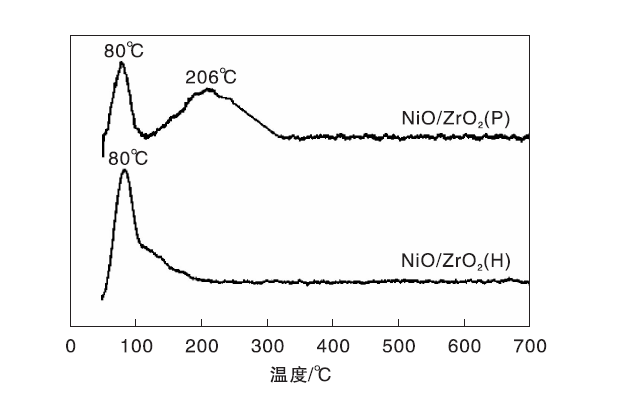 | 图6 ZrO2 (P)与ZrO2 (H)负载Ni催化剂的H2-TPD谱图Figure 6 H2-TPD profiles of ZrO2 (P) and ZrO2 (H) supported nickel catalysts |
Ni/ZrO2(P)催化剂表面具有较多的吸附活化氢位点, 在顺酐加氢反应中表现出较高的C=C键加氢活性, 但几乎没有C=O加氢活性, 即使提高反应温度或反应压力, 仍然表现出很低的C=O加氢活性。Ni/ZrO2(H)催化剂C=C键加氢活性较低, 但表现出一定的C=O加氢活性, 并且C=O加氢活性随反应温度或反应压力的提高而显著提高。表明Ni/ZrO2催化剂的C=C加氢活性与催化剂吸附活化氢的能力相关, 而催化剂的C=O加氢活性除了活性金属Ni的影响外, 还有其他因素参与并制约Ni/ZrO2催化剂的C=O加氢活性。结合本文的H2-TPR和拉曼光谱表征结果, 镍物种与ZrO2(H)载体相互作用较弱, 弱的相互作用促进Ni/ZrO2(H)催化剂表面新的氧空位产生。而ZrO2(P)载体与镍物种相互作用较强, 强的相互作用使得少量镍物种进入氧空位或ZrO2(P)载体的晶胞[14], 导致氧空位减少, 四方相结构无序化。Hu Q等[26]发现含锰尖晶石负载Cu催化剂表面氧空位能够活化丁二酸二甲酯分子中的C=O基团, 进而促进C=O加氢。本课题组的研究结果发现[14], 并不是Ni/ZrO2催化剂表面所有的氧空位均可促进丁二酸酐分子中的C=O加氢, 只有相对缺电子的氧空位才能有效地活化丁二酸酐分子中C=O基团, 进而与其邻近的Ni0协同作用完成C=O加氢, 而相对富电子的氧空位不能有效的活化丁二酸酐分子中的C=O基团, 因而未参与C=O加氢。本文不同反应温度和氢气压力条件的顺酐加氢性能评价结果证实了以上研究结果, 也进一步表明Ni/ZrO2催化剂的C=O加氢性能与载体性质有关。以ZrO2(P)为载体的Ni基催化剂无论在较高反应温度或氢气压力条件下均表现出很低的C=O加氢活性, 而以ZrO2(H)为载体的Ni基催化剂表现出较高的C=O加氢活性。结合H2-TPR和拉曼光谱表征结果, 推测Ni/ZrO2催化剂的C=O加氢活性与ZrO2载体表面结构性质有关。ZrO2载体表面结构性质不同, 与镍物种相互作用不同, 导致Ni/ZrO2催化剂呈现不同的氧空位浓度与电子性质, 从而影响Ni/ZrO2催化剂的C=O加氢性能。
3 结 论
(1)研究不同条件制备的单斜相和四方相混合晶相组成的ZrO2载体负载Ni催化剂顺酐加氢性能发现, 虽然两种ZrO2载体晶相结构组成相似, 其负载Ni催化剂在顺酐加氢反应中却表现出明显的C=O加氢性能差异。
(2)镍物种与ZrO2(H)载体相互作用较弱, 这种弱相互作用促进Ni/ZrO2(H)催化剂表面新的氧空位产生, 表面促进产生的氧空位可与其邻近的Ni0协同作用促进C=O加氢, 因而Ni/ZrO2(H)催化剂具有较高的C=O加氢活性。
(3)ZrO2(P)载体与镍物种相互作用较强, 使得少量镍物种进入氧空位或ZrO2载体晶胞, 导致Ni/ZrO2(P)催化剂表面四方相结构呈无序化, 氧空位减少, 几乎没有C=O加氢活性。
(4)通过H2-TPR和拉曼光谱表征结果可进一步看出, Ni/ZrO2催化剂表面结构性质及其C=O加氢活性与ZrO2载体的表面结构性质有关。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|