


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于间苯三酚的磺化碳基固体酸催化剂的制备及其催化油酸甲酯的合成
[张琪芳1 , 蒋平平1, *  , 李梦天
, 李梦天1 , 李晓东2 , 谢书轩2 ]
, 李梦天|
|


作者简介:张琪芳,1994年生,女,湖北省武汉市人,在读硕士研究生。
采用间苯三酚与对苯二甲醛缩聚得到的树脂为碳前驱体,分别以1,4-二氧六环与去离子水为溶剂,以溶剂热和水热法、氯磺酸为磺化试剂制备两种磺化碳基固体酸催化剂。SEM、XPS和TGA等分析表明,以1,4-二氧六环为溶剂合成的TP-A-S催化剂为形貌规整、高酸密度、良好稳定性的球形,并表现出良好的催化性能。将其用于油酸与甲醇的酯化反应,最适宜的条件为:醇油物质的量比10:1,催化剂用量占原料总质量的2.0%,反应温度70 ℃,反应时间4 h,油酸最高转化率达98.3%。且催化剂循环使用5次后,油酸转化率仍达84.4%。将制备的TP-A-S催化剂用于长链游离脂肪酸与甲醇的酯化反应,转化率高于90%,表现出良好的催化效果。
With resin from polycondensation of phloroglucinol and terephthaladehyde as precursor,using 1,4-dioxane and deionized water as different solvents,chlorosulfonic acid as sulfonated reagent,two catalysts were prepared by solvothermal and hydrothermal methods.Characterization results of SEM,XPS,TGA and so on showed that the spherical catalyst prepared by 1,4-dioxane as solvent with regular morphology,high acid density and good stability exhibited excellent catalytic avtivity.Highest oleic acid conversion reached 98.3% under the optimum esterification conditions of oleic acid and methanol which were MeOH/OA molar ratio of 10:1,catalyst dosage of 2.0% of raw material total mass,reaction temperature of 70 ℃,reaction time of 4 h.After 5 times of reuse,conversion of oleic acid was 84.4%.The prepared catalyst TP-A-S was tested in esterification of long chain free fatty acids with methanol and showed good activity,conversion was higher than 90%.
随着传统燃料的逐渐消耗与环境问题的日益恶化, 生物柴油因与柴油性能相似及其燃烧产物环保而成为化石原料的有效替代品[1]。生物柴油大多数是利用生物油脂与甲醇和乙醇等醇类进行酯化或酯交换反应得到[2, 3, 4], 其中油酸与甲醇的酯化反应是合成生物柴油的一种有效途径。
质子酸, 如硫酸、磷酸常在酯化反应中作为均相催化剂, 但其存在副反应较多、反应产生的废酸难处理且难循环使用等不足, 因此需要寻求绿色高效的催化剂代替传统催化剂[5]。近年来, 固体酸催化剂由于其可回收性和环境友好性而受到广泛关注, 其中碳基固体酸催化剂具有良好的热稳定性和催化活性, 且已被证明是一种很有前景的非均相催化剂[6, 7]。碳基固体酸催化剂研究的重点主要体现在制备碳载体和负载酸活性中心[8]两个方面。Lu W等[9]以间苯三酚和对苯二甲醛缩聚得到载体、三乙胺为胺源合成胺系多孔材料, 用于吸附脱二氧化碳, 表现出良好的性能。Kang D W等[10]以制备的酚醛树脂为原料得到的碳材料也拥有较好的性能。
本文以对苯二甲醛与间苯三酚缩聚得到的树脂为碳前驱体, 1, 4-二氧六环与去离子水为溶剂, 采用溶剂热和水热法、氯磺酸为磺化试剂制备磺化碳基固体酸催化剂, 以油酸与甲醇的酯化反应为探针, 评价其催化活性。并在室温下测定长链游离脂肪酸与甲醇酯化反应的催化活性。
间苯三酚, 纯度99%, 麦克林试剂; 甲醇、氯化钠、乙醇、1, 4-二氧六环、四氢呋喃、氢氧化钾、二氯甲烷, 均为分析纯, 国药集团化学试剂有限公司; 对苯二甲醛, 纯度98%, Meryer试剂; 氯磺酸, 纯度99%, 安耐吉试剂。
德国布鲁克公司 D8-Advance X射线衍射仪, 扫描角度为10° ~90° , 扫描速率4° · min-1, 步幅0.02° 。
德国布鲁克公司VECTOR22型傅里叶红外光谱分析仪, 试样制备采用KBr压片法。
瑞士STA409热重分析仪, 测量温度为(50~600) ℃, 升温速率10 ° · min-1。
日本日立株式会社S-4800场发射扫描电子显微镜与能谱仪。
美国赛默飞世尔科技公司X射线光电子能谱分析。
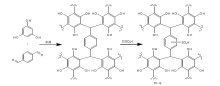
1.2.1 以1, 4-二氧六环为溶剂制备载体TP-A[11]
在25 mL的1, 4-二氧六环溶液中溶解2.522 g间苯三酚和5.365 g对苯二甲醛, 然后在70 ℃磁力搅拌1 h, 溶液变为墨绿色。将所得溶液在100 mL高温反应釜中220 ℃反应4天, 得到红褐色的固体混合物。然后过滤、四氢呋喃多次洗涤, 再将固体于50 ℃干燥12 h, 产率为92%。
1.2.2 以去离子水为溶剂制备载体TP-B
TP-B的制备与TP-A相同, 将溶剂1, 4-二氧六环替换成同体积的去离子水。
1.2.3 制备TP-A-S催化剂
将0.4 g的载体TP-A分散于50 mL二氯甲烷中, 室温下磁力搅拌1 h。在冰水浴条件下, 用漏斗滴加5 mL氯磺酸, 室温下搅拌4天。将反应后的样品稀释、过滤, 并用去离子水和甲醇洗涤至中性, 将所得黑色固体于80 ℃干燥12 h。
1.2.4 制备TP-B-S催化剂
TP-B-S与TP-A-S催化剂的合成步骤相同, 将TP-A载体替换成TP-B载体即可, 得到棕色固体。
基于间苯三酚的磺化碳基固体酸催化剂(TP-S)的合成示意图如图1所示。
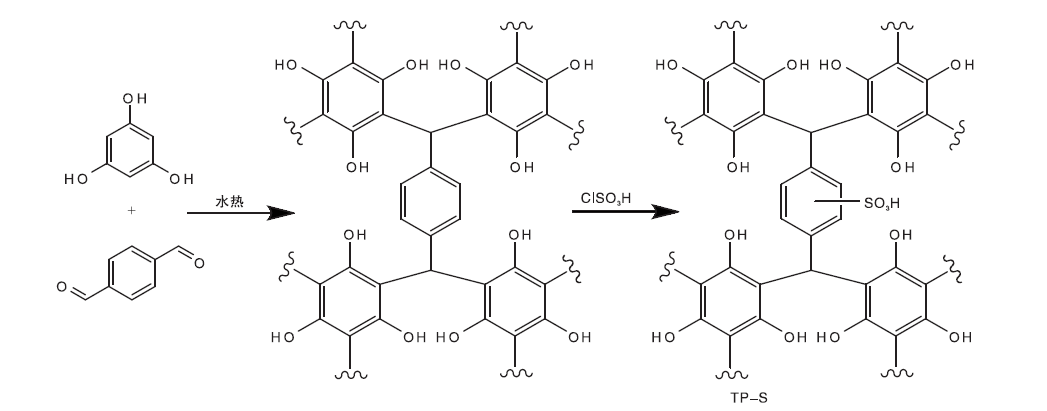 | 图1 催化剂TP-S的合成示意图Figure 1 Synthesis of the catalyst TP-S |
图2为载体TP-A与TP-B、催化剂TP-A-S与TP-B-S的SEM照片。由图2可看到, 合成的球形载体TP-B的形貌较为规整、表面平滑(e、f)。负载后的催化剂TP-B-S基本没有形变(g、h), 但球体表面出现较为明显的凸起负载物(h)。对比图2a、b可以看到, 合成的载体TP-A勉强保持了球形的轮廓, 但出现较明显的团聚, 负载后的TP-A-S催化剂表面显现较多的负载物。表明不同种类的溶剂对载体表面形貌有较为显著的影响。
 | 图2 载体及催化剂的SEM照片 a、b.TP-A; c、d.TP-A-S; e、f.TP-B; g、h.TP-B-SFigure 2 SEM images of carriers and catalysts |
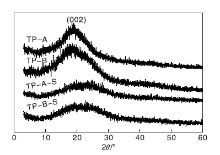
图3为载体TP-A与TP-B、催化剂TP-A-S与TP-B-S的XRD图。从图3可以看出, 在10° ~30° (002)载体TP-A和TP-B均有一个较宽的信号峰, 且没有较大的区别, 表明溶剂对载体结构没有明显影响, 合成的载体为无定形碳[13]。而TP-A-S和TP-B-S催化剂的出峰位置和载体TP-A与TP-B相近, 但峰强度有所减小, 表明活性中心的负载对催化剂结构有一定程度的影响。
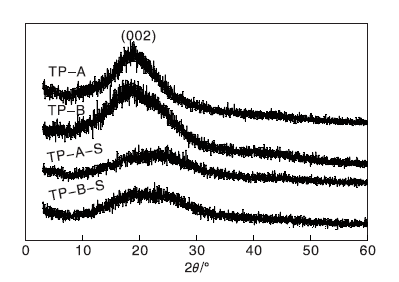 | 图3 载体及催化剂的XRD图Figure 3 XRD patterns of carriers and catalysts |
图4为载体TP-A与TP-B、催化剂TP-A-S与TP-B-S的全元素扫描XPS谱图。从图4可知, 相对于载体TP-A和TP-B, 催化剂TP-A-S和TP-B-S的CKLL、OKLL俄歇峰没有明显移动, 但出现S2s和S2p两个新的峰。在TP-A-S和TP-B-S催化剂S2p窄谱图中可以看到, 169.1 eV处出现一个明显的峰, 归属于硫的氧化物(-C-SOx-)[12], 表明载体上成功负载了-SO3H, 也证明图2d、h照片上的凸起负载物为磺酸基团。
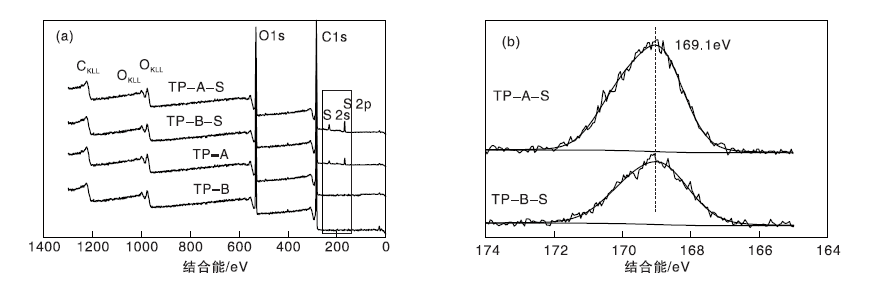 | 图4 载体及催化剂的全元素扫描XPS谱图Figure 4 XPS spectra of carriers and catalysts |
图5为TP-A-S和TP-B-S催化剂的EDS图。对比图5可知, 两种催化剂上C、O和S元素分布均匀且含量较多, Cl元素的含量较少。
 | 图5 TP-A-S和TP-B-S催化剂的EDS图 a、h.背景(a为TP-A-S, h为TP-B-S); b、i.C元素; c、j.O元素; d、k.S元素; e、l.Cl元素Figure 3 EDS images of TP-A-S and TP-B-S catalysts |
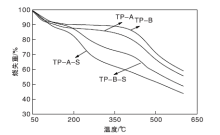
图6为载体TP-A与TP-B、催化剂TP-A-S与TP-B-S的热重曲线。
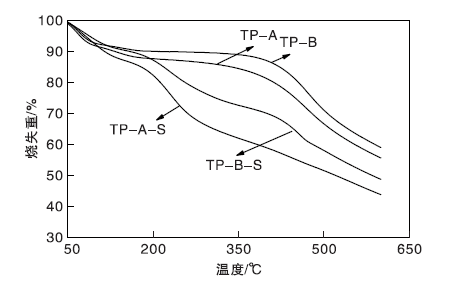 | 图6 载体及催化剂的热重曲线Figure 6 TGA curves of carriers and catalysts |
由图6可以看出, 低于100 ℃的失重是样品表面水分的脱除。载体TP-A和TP-B在高于400 ℃的明显失重是碳材料的氧化分解所致。催化剂TP-A-S和TP-B-S在高于200 ℃的失重是由于磺酸基团脱落和碳载体的氧化分解[15, 16], 表明催化剂TP-A-S和TP-B-S在低于200 ℃的热力学性质相对稳定[17]。
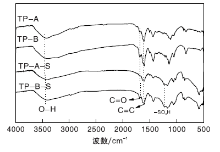
载体TP-A与TP-B、催化剂TP-A-S与TP-B-S的FT-IR谱图如图7所示。由图7可以看出, 与载体TP-A和TP-B相比, TP-A-S和TP-B-S催化剂在1 224 cm-1处均出现-SO3H伸缩振动峰[14], 表明-SO3H成功负载于碳球TP-A、TP-B上。
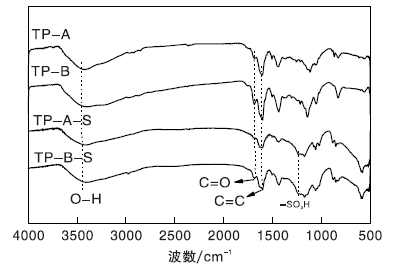 | 图7 载体及催化剂的FT-IR谱图Figure 7 FT-IR spectra of carriers and catalysts |
采用间接滴定法测定[18]TP-A-S和TP-B-S催化剂的总酸量。测定过程如下:在20 mL、0.1 mol· L-1的NaCl溶液中加入0.15 g催化剂, 磁力搅拌24 h, 过滤, 用0.01 mol· L-1的NaOH溶液滴定滤液至中性, 计算得到催化剂样品的总酸量。TP-A-S催化剂总酸量为2.26 mmol· g-1, TP-B-S催化剂总酸量为2.13 mmol· g-1。
将一定比例的油酸与甲醇加至100 mL烧瓶中, 加热回流并搅拌。反应中每隔1 h取样, 测定反应体系的酸值[19]并计算反应物油酸转化率[20]。反应前体系的初始酸值为178 mg-KOH· g-1。
2.9.1 催化剂种类
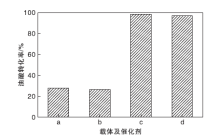
图8为载体TP-A与TP-B、催化剂TP-A-S与TP-B-S在油酸甲酯合成反应中的催化活性。从图8可以看到, 以载体TP-A和TP-B为催化剂时, 油酸转化率分别为28.1%和26.5%, 表明合成的载体对反应也有微小的催化活性。而负载活性中心后, TP-A-S和TP-B-S催化剂上油酸转化率分别可达98.3%和96.8%, TP-A-S比TP-B-S催化剂的催化效果更好, 这是由于TP-A-S催化剂拥有更高的总酸量。为使研究更加具有针对性, 以下实验均使用TP-A-S催化剂进行条件优化。
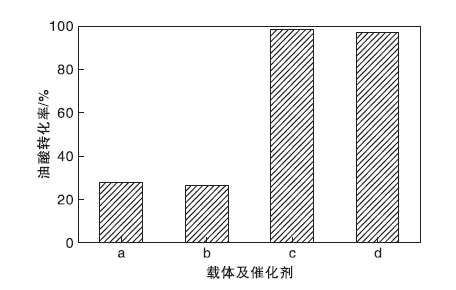 | 图8 不同载体及催化剂上油酸转化率 a.TP-A; b.TP-B; c.TP-A-S; d.TP-B-SFigure 8 Conversion of oleic acid over different carriers and catalysts |
2.9.2 反应时间
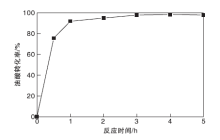
图9为不同反应时间对油酸转化率的影响。从图9可以看出, 反应时间为0.5 h时, 油酸转化率为75.5%; 反应时间4 h时, 油酸转化率最高, 为98.3%, 继续增加反应时间, 转化率变化不明显, 选择佳反应时间为4 h。
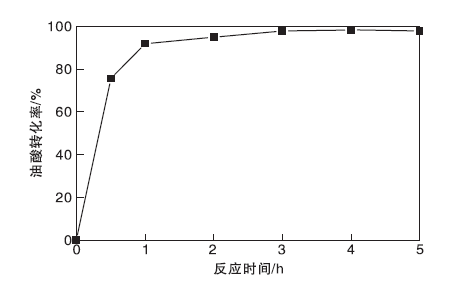 | 图9 反应时间对油酸转化率的影响Figure 9 Effect of reaction time on conversion of oleic acid |
2.9.3 反应温度
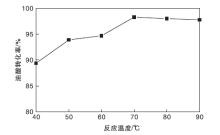
图10为反应温度对油酸转化率的影响。由于酯化反应为吸热反应, 在一定温度范围, 反应会伴随温度的增加而正向移动, 油酸转化率会逐渐提高。由图10可知, 在反应温度40 ℃时, 油酸转化率为89.4%, 表明TP-A-S催化剂在低温下也具有较强的催化活性。当反应温度增加到70 ℃时, 油酸转化率最高, 为98.3%。继续升高反应温度, 油酸转化率略有下降, 选择最佳反应温度为70 ℃。
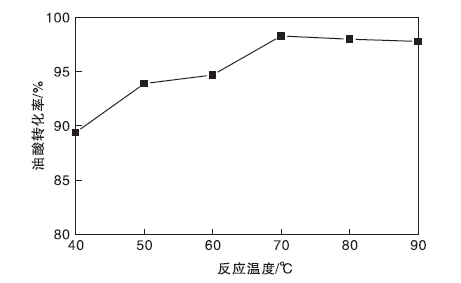 | 图10 反应温度对油酸转化率的影响Figure 10 Effect of reaction temperature on oleic acid conversion |
2.9.4 醇油物质的量比
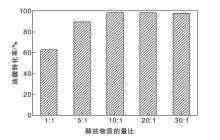
图11为醇油物质的量比对油酸转化率的影响。由图11可以看出, 当醇油物质的量比为1:1和5:1时, 油酸转化率低于90%, 表明反应不完全。当醇油物质的量比增大到10:1时, 油酸转化率提高至98.3%。这是由于该反应为可逆反应, 当增大反应物甲醇的浓度时, 反应会正向移动, 促使油酸转化率增加。而进一步增大醇油物质的量比时, 油酸转化率变化甚微, 表明当醇油物质的量比为10:1时, 反应已基本达到平衡。因此最佳醇油物质的量比为10:1, 既可保证高油酸转化率, 又体现出原料的最大化利用。
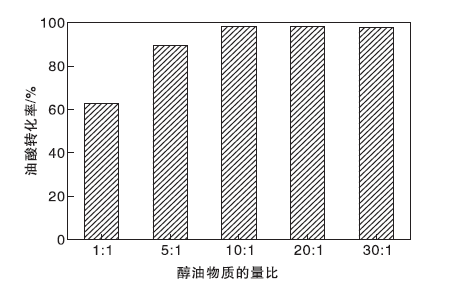 | 图11 醇油物质的量比对油酸转化率的影响Figure 11 Effect of n(MeOH):n(OA) on conversion of oleic acid |
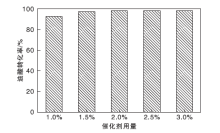
2.9.5 催化剂用量
催化剂用量对油酸转化率的影响如图12所示。从图12可以看到, 当催化剂用量增加时(1.0%增至2.0%), 油酸转化率提高较明显, 最高达98.3%。这是因为体系中的酸活性位点有所增多, 催化活性提升。而继续增加催化剂用量, 油酸转化率保持平稳, 选择最佳催化剂用量为原料总质量的2.0%。
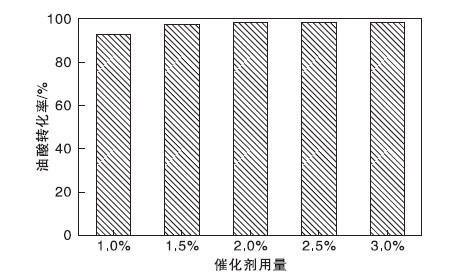 | 图12 催化剂用量对油酸转化率的影响Figure 12 Effect of catalyst dosage on conversion of oleic acid |
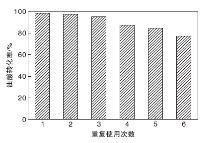
2.9.6 催化剂重复使用性能
在醇油物质的量比10:1、催化剂用量占原料总质量的2.0%、反应温度70 ℃、反应时间4 h条件下, 对TP-A-S催化剂的稳定性进行测试。反应完成后, 离心分离催化剂并使用乙醇多次洗涤, 在80 ℃干燥过夜。干燥后的催化剂直接用于下一次反应, 结果如图13所示。由图13可以看出, TP-A-S催化剂重复使用5次后, 油酸转化率仍能达到84.4%, 表明催化剂TP-A-S的稳定性良好。测得第5次重复使用的TP-A-S催化剂表面酸量为1.74 mmol· g-1, 这可能是由于在多次试验及洗涤过程中, 部分活性中心-SO3H脱落, 导致催化剂活性降低。
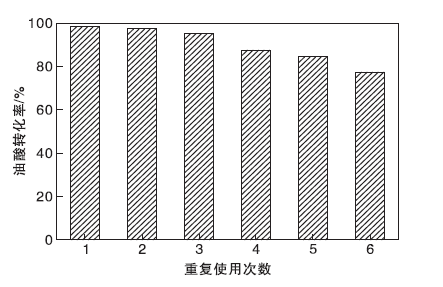 | 图13 催化剂重复使用次数对油酸转化率的影响Figure 13 Effect of catalyt reused times on conversion of oleic acid |
(1) 采间苯三酚和对苯二甲醛缩聚得到的树脂TP-A和TP-B为载体, 以溶剂热和水热法、氯磺酸为磺化试剂制备的催化剂TP-A-S和TP-B-S均为形貌相对规整的球体, 具有较高的总酸量(2.26 mmol· g-1、2.13 mmol· g-1)和良好的热稳定性。
(2) 将磺化碳基固体酸催化剂TP-A-S和TP-B-S用于合成油酸甲酯的酯化反应, 结果表明, TP-A-S催化剂催化活性更为优异, 在醇油物质的量比为10:1、催化剂用量为原料总质量的2.0%、反应温度70 ℃条件下反应4 h, 油酸转化率最高可达到98.3%。且TP-A-S催化剂在重复使用5次后, 油酸转化率仍可达到84.4%, 具有良好的循环使用性。
(3) 将制备的TP-A-S催化剂用于长链游离脂肪酸与甲醇的酯化反应, 转化率高于90%, 表现出良好催化效果。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|