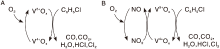
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
含氯挥发性有机物的催化氧化研究进展
[蒋熙云, 杨军, 刘雨溪, 邓积光, 敬林, 戴洪兴*  ]
]
]
|
|


催化氧化是控制挥发性有机污染物排放的有效途径之一,但传统催化剂在氧化消除含氯挥发性有机物(CVOCs)时易中毒而失活,因此,研发新型、高效、稳定的催化剂对其工业应用具有重要意义。简要综述近年来稀土及过渡金属氧化物、分子筛和负载贵金属催化剂对CVOCs氧化的催化性能和催化反应机理,剖析CVOCs氧化反应副产物的形成和催化剂失活的原因,并展望CVOCs催化氧化技术的未来发展趋势。
Catalytic oxidation is one of the most effective pathways in controlling the emission of volatile organic compouns,but the conventional catalysts are prone to be deactivated due to chlorine poisoning during the oxidation processes of chlorinated volatile organic compounds(CVOCs).Therefore,the development of novel,high-efficiency,and durable catalysts is of significance in industrial applications.In this review,we briefly summarize the recent progress in catalytic performance and reaction mechanisms of rare earth oxides,transition metal oxides,zeolites,and supported noble metals in the oxidation of CVOCs.The formation of by-products and the reasons of catalyst deactivation in CVOCs oxidation are discussed.In addition,the development trend of the CVOCs oxidation technology in the future is envisioned.
大部分挥发性有机化合物(VOCs)污染大气环境, 危害人体健康, 破坏生态平衡, 因此, 必须严格控制VOCs污染物排放, 改善大气环境, 保持人类社会可持续发展。常见的 VOCs包括烷烃、烯烃、卤代烃、醇、醛、羧酸和酯等, 含氯挥发性有机物(CVOCs)为氯代烷烃和氯代烯烃、氯代芳香烃等, 主要包括二氯甲烷、四氯化碳、1, 2, 4-三氯苯、三氯乙烯、氯苯、1, 2-二氯苯和1, 2-二氯乙烷等, 污染源包括医药、印刷、橡胶和农药等行业。多数CVOCs具有良好的化学稳定性和热稳定性, 不易分解或生物降解, 可在自然界中长时间滞留, 对环境造成持久性污染。CVOCs因其高毒性、反应惰性和容易使催化剂中毒而成为较难处理的一类有害物质。
催化氧化法因具有起燃温度低、处理效率高以及不易生成二次污染等优点而被认为是最具前景的CVOCs消除技术, 然而在CVOCs氧化过程中产生的HCl和Cl2可使催化剂中毒而失活, 影响其消除效果。因此, 研发新型高效稳定的催化剂具有重要的实用价值。CVOCs的催化氧化研究开始于20世纪70年代, Pt/Al2O3和13X型分子筛是最早应用于CVOCs氧化的催化剂[1, 2]。90年代人们发现二噁英、多氯联苯等持久性有机污染物对人体和环境造成严重危害, 研发了负载贵金属[3, 4, 5]、金属氧化物[6, 7, 8]和固体酸[9, 10]等催化剂, 用于CVOCs的催化氧化。其中, Hutchings G J研究小组[7, 11]报道的铀基催化剂U3O8可以在350 ℃将浓度为 10 000× 10-6和空速为70 000 h-1的氯苯完全氧化, 并在400 h内催化活性保持稳定, 但铀化合物具有放射毒性, 不易实现工业应用。虽然贵金属Pt和Pd的催化活性较好, 但价格昂贵, 对CVOC分子的氯取代反应活性高于氧化反应活性, 导致高氯代副产物的产生[5, 12], 且这类副产物的毒性大于反应物本身。近年来, CVOCs氧化催化剂的研究集中在Ce、Mn、Cr、V、Co等过渡金属氧化物或其复合氧化物方面, 研发高活性、高抗氯中毒能力的催化剂是CVOCs催化氧化技术的关键。
已有一些有关CVOCs氧化消除的文献综述报道
CVOCs氧化催化剂可分为稀土及过渡金属氧化物催化剂、分子筛催化剂和负载贵金属催化剂, 表1列出一些典型CVOCs氧化催化剂的催化活性。
| 表1 用于CVOCs氧化的典型催化剂的催化活性 Table 1 Catalytic activities of typical catalysts for the oxidation of CVOCs |
Ce、Mn、Cr、V、Co等金属氧化物及其复合氧化物对CVOCs催化氧化表现出优异的活性, 将其作为活性组分负载于TiO2、ZrO2、Al2O3、分子筛等高比表面积的载体上, 获得活性组分呈分散态的催化剂, 进一步改善催化性能。
1.1.1 Cr基催化剂
Liu J D等[45]研究了尖晶石型CoCr2O4催化剂对二氯甲烷氧化的催化性能, 根据计算的转换频率(TOFs)可知, 高价态的Cr6+物种比Cr3+具有更高的催化活性, 经600 ℃焙烧处理, 使部分Cr3+和Cr6+取代位于催化剂表面八面体空位中的Co3+, 表面暴露更多的Cr6+物种, 同时提高催化剂的表面酸性, 从而显著改善催化剂催化性能。并且二氯甲烷氧化的单位比表面积反应速率与催化剂酸性、氧化还原能力以及Cr6+浓度呈现出正相关性, 催化剂的高活性是这些因素协同作用的结果。
Zhou R X课题组[46, 47, 48, 49, 50, 51, 52, 53]制备出系列铈铬复合金属氧化物催化剂, 通过研究其对二氯乙烯和三氯乙烯氧化的催化活性, 发现这些催化剂较高的活性取决于其较大的比表面积、较多的酸性位和较强的氧化还原能力。研究认为, 大量具有强氧化能力的Cr6+物种产生于CeO2与CrOx之间的强相互作用, 有利于反应物和中间产物的深度氧化。选择具有丰富酸性位的HZSM-5、USY、Nb2O5等固体酸为载体所制备的催化剂催化活性较高, 原因是其中的酸性位和氧化还原中心产生了协同作用, 但是水蒸汽的引入使催化剂催化活性略有下降, 同时副产物的形成也有所减少。
1.1.2 V基催化剂
V2O5/TiO2催化剂普遍用于工业废气的氮氧化物选择性催化还原(SCR), 加入W、Mo等助剂可提升催化活性和抗硫性能。此类催化材料对CVOCs尤其是氯代芳香烃的催化氧化同样具有良好的催化活性和稳定性。对氯苯类化合物的催化氧化反应, V催化剂相较于贵金属催化剂具有更少的氯代副产物和更高的HCl选择性, 其催化活性随着氯苯氯化程度的升高而下降[54]。
Hetrick C E等[55]发现水蒸汽在不同温度范围对V2O5/TiO2催化剂上二氯苯氧化反应的影响存在相反趋势。当温度低于270 ℃时, 水蒸汽可促进表面Cl和C物种的移除, 但温度高于270 ℃时, 水蒸汽对活性位的竞争吸附导致催化活性下降。Gaigneaux E M课题组[56, 57, 58, 59, 60]系统研究了氯苯类化合物在V2O5/TiO2催化剂上的氧化反应, 发现NO的引入可提高氯苯转化率。氯苯在VOx上催化氧化反应遵循典型的Mars-van Krevelen机理(图1), 引入NO后, NO在WOx、MoOx等助剂表面被氧化成NO2, NO2再氧化被部分还原的V物种。NO2在V4+Ox« V5+Ox的氧化还原循环中比O2更为高效, 因此催化活性显著提升。并且发现, 采用Ce掺杂或S
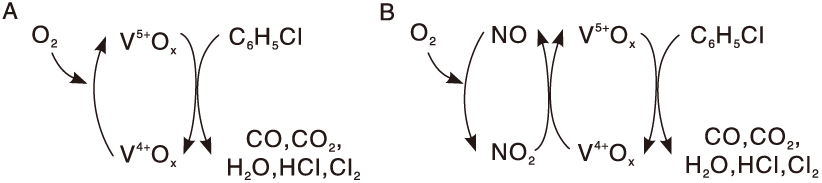 | 图1 NO的引入对氯苯在VOx上氧化反应机理的影响示意图[56]Figure 1 Scheme of chlorobenzene oxidation over the VOx catalyst in the absence or presence of NO[56] |
Wang J等[61]研究了V2O5/TiO2对氯苯和多氯苯二元混合CVOCs催化氧化反应, 发现与单一反应物体系相比, 在多氯苯存在时氯苯转化率下降, 这是由于催化剂更易吸附多氯苯而发生亲核取代反应, 低温竞争吸附抑制了氯苯转化。随着反应温度上升, 多氯苯转化率低于氯苯转化率, 归因于在高温时内扩散对反应影响增大, 多氯苯较大的分子直径使其具有较大的内扩散阻力。
此外, Wang X Y课题组[62, 63]以高活性的CeO2为载体制备负载VOx催化剂, 研究其对二氯乙烯、二氯甲烷和氯苯氧化的活性和稳定性, 发现无定形的VOx高分散于CeO2表面, 丰富的L酸位有利于CVOCs分子的吸附。二氯乙烯氧化产物中含有较多的CO, 这是由于VOx表面晶格氧的氧化能力较弱, 难以将中间产物(乙醛)完全氧化为CO2所致。
1.1.3 Mn基催化剂
Mn具有多种氧化态, 其氧化物主要有MnO、Mn3O4、Mn2O3和MnO2等。氧化锰在催化过程中易发生氯化反应, 生成氧氯化物导致催化剂催化活性下降。
Liu Y等[64]将锰氧化物负载到不同载体上考察对氯苯氧化的活性, 结果表明, SiO2表面难以高分散MnOx物种, MnOx和Al2O3载体之间的相互作用太强而导致MnOx物种的氧化还原能力较弱, TiO2表面的MnOx物种具有最好的氧化还原性能, 因而催化活性最高。还制备了TiO2改性的Al2O3复合载体, 再采用浸渍法制备负载氧化锰催化剂, 观察到空速为8 000 h-1时, 氯苯完全转化温度为300 ℃[65]。Vu V H等[66]制备的MnCuOx/TiO2催化剂在350 ℃即可使氯苯完全氧化, 连续反应5天后, 催化活性无明显下降; 但在300 ℃时, 氯苯转化率在5 h下降至75%, 这是由于氯苯中的Cl与活性组分反应生成氧氯物种使催化剂失活所致。Weng X L等[67]采用水热法制备不同形貌和晶型的g-MnO2、a-MnO2和d-MnO2催化剂, 由H2-TPR和O2-TPD表征结果可知, 具有层状结构的d-MnO2具有最好的氧化还原性能, 对氯苯催化氧化活性最高, 但CO2选择性最低, 这是由于d-MnO2催化剂表面具有最多的Mn4+物种, 在反应过程中易生成Mn-Cl键, 阻碍Cl的脱除, 导致催化剂表面富集氯而失活, CO2选择性差。
1.1.4 Fe基催化剂
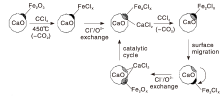
Fe基催化剂虽然廉价且易制备, 但对CVOCs催化氧化的温度较高。Decker S P等[68]将Fe2O3负载于CaO纳米晶表面并用于CCl4的催化消除, 发现在高温下CCl4与氧化铁反应生成FeClx, 而FeClx和CaO之间存在Cl-/O2-离子交换(图2), 可使氯化后的FeOx再生, 而氯离子可在CaO表面迁移直至达到饱和。
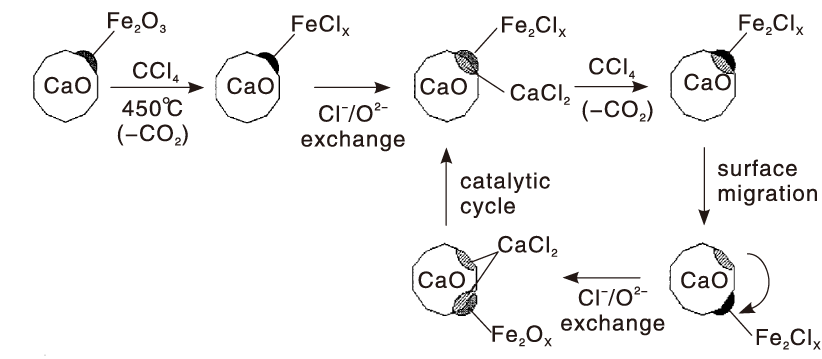 | 图2 CaO负载Fe2O3催化剂降解CCl4的Cl转移机理[68]Figure 2 Cl transfer mechanism for the degradation of CCl4 over CaO-supported Fe2O3 catalyst[68] |
Ma X D等[69, 70, 71]将CaCO3或CaO引入a-Fe2O3中制成复合氧化物, 并用于1, 2-二氯苯的氧化, 发现CaCO3或CaO助剂与Fe2O3之间的强相互作用提高了催化剂催化活性, 将其负载于TiO2载体上, 催化氧化活性进一步改善, 且抗水性能和抗氯中毒能力提高。
1.1.5 Ce基催化剂
CeO2晶胞中存在Ce3+ « Ce4+的氧化还原循环, 具有良好的储释氧性能。用于CVOCs氧化的Ce基催化剂主要包括Ce
de Rivas B等[79]在室温下采用盐酸浸渍法经在空气中焙烧制得氯化处理的Ce-Zr复合氧化物催化剂, 发现氯化对催化剂的二氯乙烷氧化活性几乎没有影响。一方面氯化处理破坏了催化剂的孔结构, 比表面积下降; 另一方面C
Sun P F等[31]研究了K改性的MnCeO/HZSM-5催化剂对含氯芳烃氧化的催化性能, 发现K掺杂增加催化剂的L酸性和亲水性, 改善催化剂的深度氧化能力和水解反应能力, 从而显著提高CO2和HCl选择性。
1.1.6 钙钛矿型复合金属氧化物催化剂
钙钛矿型氧化物(ABO3)价格低廉, 抗Cl中毒性能较好, 但氧化性和酸性较弱, A位或B位元素被其他元素取代后可增加晶格缺陷, 提升氧化还原能力, 在CVOCs催化氧化领域具有潜在的应用前景。
Zhang C H等[82]利用Sr、Mg、Ce部分取代LaMnO3中的La并评价其对氯乙烯氧化的催化活性, 观察到Mg或Ce的部分取代提高了催化剂催化活性, 而Sr的部分取代降低了催化剂催化活性。活性最高的La0.8Ce0.2MnO3催化剂在空速15 000
分子筛是一类具有特定孔道结构和丰富酸性中心的多孔材料, 表面酸中心为CVOCs吸附和活化的活性位点, 因此, 分子筛对CVOCs的催化氧化得到广泛研究。
Scirè S[85]比较了以γ -Al2O3和HZSM-5与H-beta两种分子筛为载体负载质量分数0.5%Pt催化剂对氯苯氧化的催化性能, 发现催化剂的活性为Pt/H-ZSM-5 > Pt/H-beta > Pt/γ -Al2O3, 多氯苯副产物PhClx的生成量与此顺序正好相反。Pt/H-ZSM-5催化剂催化活性最高, 是由于H-ZSM-5分子筛具有最强的表面酸性, 有利于氯苯的吸附, 且HZSM-5分子筛在反应中表现出明显的择形效应, 其较小的孔径限制了PhClx分子进入孔道内部, 从而减少多氯苯的形成。在研究了H-ZSM-5、H-Y分子筛对二氯乙烯和三氯乙烯的催化氧化性能后, Ló pez-Fonseca R研究小组[10, 86]发现, H-ZSM-5分子筛具有较强的B酸位, 使其活性高于H-Y分子筛, H-Y分子筛对HCl和CO2选择性更高。H-Y分子筛经脱铝改性后, 虽然强的B酸位量减少, 但其他酸位强度增加, 催化活性提高。引入H2O后, H2O与反应物分子在活性位上产生竞争吸附, 导致活性下降。单组分分子筛表面的活性氧物种较少, 反应产物中有大量CO形成。通过离子交换法对分子筛进行贵金属掺杂改性后[44, 87, 88], 催化活性和CO2选择性大幅增加, 同时Cl2和多氯代副产物数量提高, 但H2O的引入可减少氯代副产物的生成并提高催化活性。Aranzabal A等[89]比较了不同类型的分子筛(H-ZSM-5、H-MOR、H-BEA)催化氧化二氯乙烯和三氯乙烯的稳定性, 结果表明, 分子筛表面强B酸位上的积炭和氯吸附是导致催化剂失活的主要原因。具有一维孔道结构的H-MOR分子筛的积炭最严重, 而H-ZSM-5分子筛因其三维连通的孔结构和较高的硅铝比而具有最好的稳定性。H2O的引入可抑制积炭和Cl中毒, 维持催化剂的酸性和活性, 从而避免催化剂失活。
Pt、Ru、Pd、Rh等贵金属对C— C、C— H、C— O键具有较强的活化能力, 在催化VOCs氧化反应中表现出优异的低温活性和良好的稳定性。然而, 在CVOCs氧化反应中, 贵金属在低温下与Cl强烈作用, 覆盖了活性位, 导致低温活性下降。这一缺点阻碍了贵金属催化剂在CVOCs氧化反应中的广泛应用。
1.3.1 负载Pt催化剂
van den Brink R W等[5]研究了Pt/g-Al2O3催化剂对氯苯氧化反应的性能, 发现在440 ℃条件下, 氯苯完全转化后有大量多氯苯副产物生成, 主要是Pt和Cl生成的PtOCl催化吸附的氯苯继续氯化, 向体系中引入水蒸汽可减少多氯苯的产生, 从而提高催化活性。氯苯在g-Al2O3上的完全转化温度为550 ℃, 但没有多氯苯的形成。并研究了焙烧温度对Pt/γ -Al2O3催化剂催化氧化氯苯性能的影响[12], 结果显示, 粒径小的Pt粒子更容易生成PtOCl物种, 高温焙烧所得催化剂因具有较大粒径的Pt粒子而使多氯苯的生成大幅减少。低的O2浓度也对副产物的减少起到促进作用。此外, 在反应气中引入烷烃可有效减少高氯代副产物的形成[90], 提高氯苯完全氧化活性。主要原因是烃类化合物能将Pt还原到活性最高的零价态, 抑制氧氯化铂物种生成, 促进催化剂表面Cl移除, 从而改善催化剂的稳定性。还利用同位素示踪分子对比了Pt/Al2O3催化剂上苯与氯苯的催化氧化过程以及C6H5Cl和C6D5Cl在Pt/Al2O3催化剂上的反应性差异[91], 未发现由于H/D差异而产生的同位素效应。当氯苯吸附时, C— Cl键断裂形成的氯吸附在Pt上, 导致两者活性无变化, 但在反应气中引入过量正己烷后, 表面Cl可顺利移除, 此时C— H键的断裂成为速控步骤, C6H5Cl和C6D5Cl的反应速率常数体现出明显的同位素效应。
Pitkä aho S等[26]分别将Pt、Pd、Rh、V2O5等活性组分负载到Al2O3载体上, 考察其在潮湿气氛下对二氯甲烷氧化的催化活性, 结果表明, 二氯甲烷在Pt/Al2O3和Rh/Al2O3催化剂上完全转化温度分别为420 ℃和440 ℃。利用CeO2对Al2O3载体进行改性, 对产物选择性产生积极影响, HCl选择性提高, CO选择性降低, 但氯甲烷转化率没有显著改变。经过40 h连续反应, 催化活性无明显下降, 催化剂表面也没有明显的积炭。
1.3.2 负载Pd催化剂
对VOCs氧化反应, 贵金属中Pd对C3以下的烃类表现出最好的活性, 而对长链烃氧化反应Pt的活性更高。对CVOCs而言, Pt对氯苯氧化的催化活性高于Pd, 而Pd对氯化反应的活性高于其氧化活性[12]。对于二氯乙烯和三氯乙烯氧化, 尽管Pt表现出更高的CO2选择性, 但Pd的活性更好[92]。
Giraudon J M等[93]研究了TiO2、ZrO2负载Pd催化剂对氯苯氧化的活性, 结果表明, Pd/TiO2催化剂催化活性更高, 这是由于TiO2载体具有更好的还原性(Ti4+ ® Ti3+)。Becker L等[4]制备了酸改性的沸石负载Pd催化剂用于氯苯催化氧化, 向沸石晶格中引入质子可加速Cl以HCl的形式脱除, 从而减少多氯苯副产物的生成, 但由此消耗的质子无法通过原料气补充。增加O2浓度也对降低副产物有积极作用, 这是由于过量的氧占据了氯化反应的活性位所致。
1.3.3 负载Ru催化剂
Ru价格低于其他贵金属, 对Deacon反应具有优良的催化活性, 抗Cl中毒能力较强, 已得到工业应用[94]。
据Miranda B等[95]报道, Al2O3负载Ru催化剂相较于负载Pt、Rh、Pd催化剂, 在三氯乙烯氧化反应中表现出更好的催化活性和稳定性, 但副产物分布截然不同, 认为三氯乙烯在Ru催化剂上氧化反应机理与在其他3种贵金属催化剂上的各异。Cao S等[96]考察了介孔TiO2负载不同贵金属催化剂对二氯甲烷氧化的催化性能, 发现Pt/TiO2催化剂催化活性最差且最易失活, 虽然Pd/TiO2催化剂的初始活性最高, 但反应6 h后催化活性和CO2选择性均下降; 在Ru/TiO2催化剂上产生的C和Cl物种能够快速被移除, 避免了积炭和氯中毒, 因而Ru/TiO2催化剂的稳定性最好。Ru与载体之间的相互作用是影响CVOCs催化氧化性能的重要因素。Huang H等[32]采用水热法制备棒状、立方块状和八面体状CeO2纳米粒子, 以此为载体制备负载Ru催化剂, 并研究其对氯苯氧化的催化活性, 发现棒状CeO2(CeO2-r)优先暴露(100)和(110)晶面, 在这些晶面上负载的Ru形成大量的Ru-O-Ce键, 使Ru/CeO2-r具有最高的Ru4+浓度和最好的表面氧迁移性, 因而CeO2-r对氯苯氧化的催化活性最高。
CVOCs的催化氧化反应是一个复杂过程, 反应的第一步为CVOCs分子在活性位上的解离吸附。由于C— Cl键的键能比C— H和C— C键低, 故发生解离的通常是C— Cl键。氯苯在Pd上解离吸附于室温下即可发生[97]。
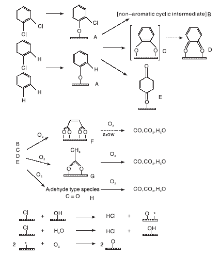
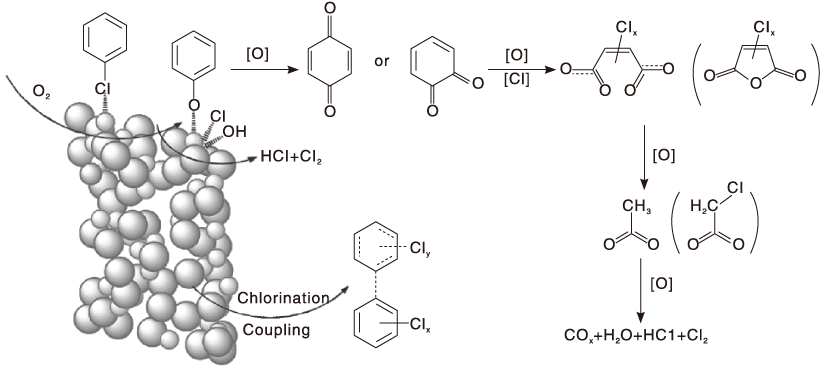
Lichtenberger J[98]利用原位红外光谱和动力学方法研究不同氯取代苯(苯、氯苯、1, 2-二氯苯、1, 3-二氯苯和1, 4-二氯苯)在V2O5/TiO2催化剂上的氧化反应, 并以氯代环己烷为对照物, 结果发现, 氯代环己烷比苯和氯代苯更易被氧化, 表明苯环的开环可能是速控步骤。氯取代基的吸电子效应使苯的吸附活化能明显高于氯代苯, 氯取代基的数量和位置也对反应动力学产生影响。并提出氯代苯在V2O5/TiO2催化剂上氧化反应机理, 如图3所示。反应物分子先经亲核取代吸附于催化剂表面, 再与较高温度下吸附在催化剂表面的部分脱氯产物发生亲电取代反应, 生成邻位、对位苯醌或非芳香族中间体。这些产物可进一步氧化生成马来酸盐、醋酸盐或甲酸盐, 最后形成COx和H2O。表面Cl则通过与表面羟基或H2O反应而被移除, 最后气态氧补充消耗的表面氧物种, 实现催化循环。
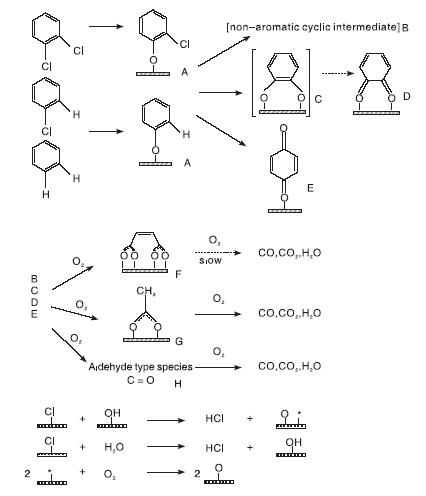 | 图3 氯苯类化合物在V2O5/TiO2催化剂上的氧化反应机理[98]Figure 3 Reaction mechanism for the oxidation of chlorinated benzenes over V2O5/TiO2 catalysts[98] |
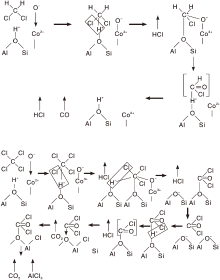
Ramachandran B等[99]研究了Co改性的Y型分子筛上二氯甲烷和四氯化碳的催化氧化反应机理, 如图4所示。二氯甲烷先吸附在催化剂表面B酸位(羟基)上, 脱去一个HCl分子形成碳正离子。O2在Co位点上解离吸附, Co2+氧化为Co3+, 同时形成
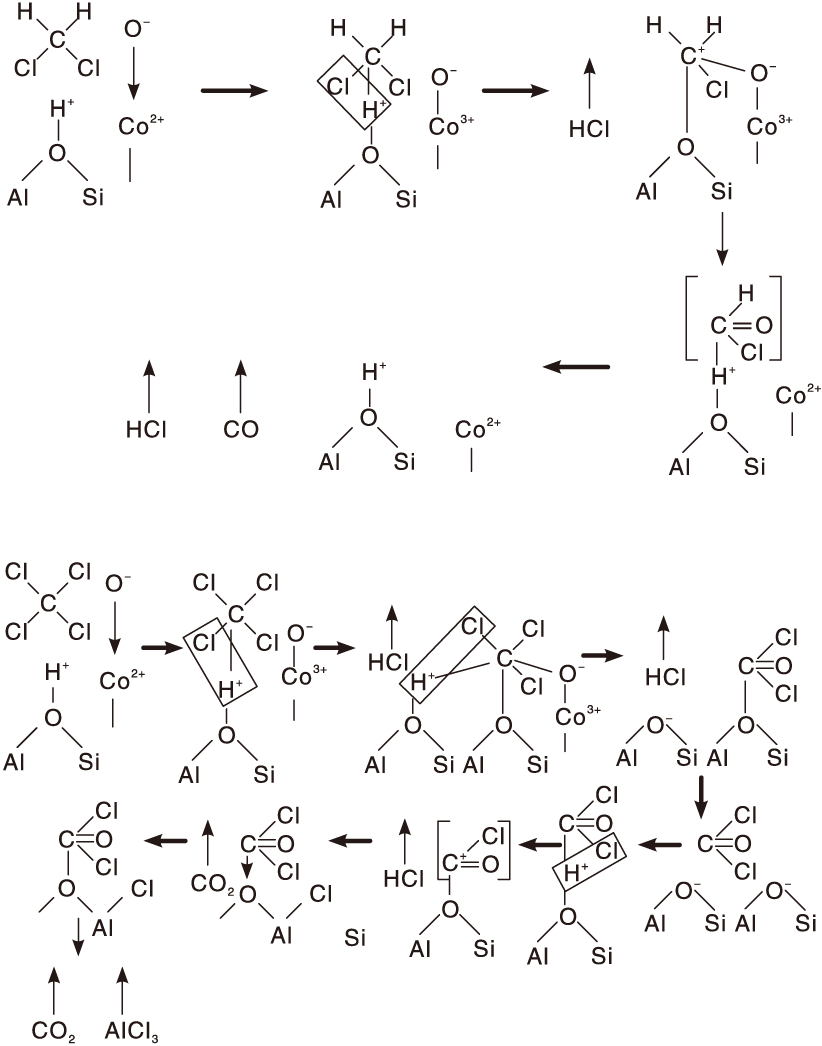 | 图4 Co-Y催化剂上二氯甲烷和四氯化碳的氧化反应机理[99]Figure 4 Reaction mechanism for methylene chloride and carbon tetrachloride oxidation[99] |
四氯化碳氧化反应机理与二氯甲烷主要区别在于在干燥条件下前者无氢源, 导致催化剂B 酸位的羟基无法恢复, 催化剂骨架易破裂。四氯化碳先在催化剂上解离脱去HCl生成C+Cl3碳正离子, C+Cl3物种再与另一B酸位羟基结合脱氯形成CCl4H+, 再与阳离子位上吸附氧反应生成光气和HCl。生成的光气分子重新吸附到附近B酸位上形成COCl+, 活性COCl+物种使沸石中的铝氯化, 并夺走骨架中的O形成CO2。
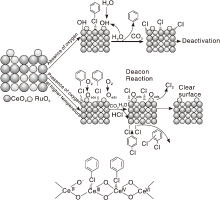
Dai Q G等[100]利用程序升温表面反应技术研究CeO2和掺杂Ru的CeO2催化剂上氯苯氧化反应机理, 结果见图5。Ce3+/Ce4+为氧化反应的活性位, 其L酸位较好地吸附氯苯分子, 通过亲核反应使氯苯解离, 产生的芳香基团进一步被表面活性氧物种氧化。尽管掺杂的Ru覆盖了部分Ce3+/Ce4+活性位, 但作为Deacon反应活性位, 将表面Cl物种快速氧化成Cl2, 避免活性位被Cl中毒, 从而提高催化剂稳定性。少量气态氯苯在部分氯化的Ru和Ce位点上通过亲电取代生成1, 2-二氯苯和1, 4-二氯苯等副产物。无氧条件下, 尾气中除CO2还产生了少量苯。此外, 在原料气中引入H2O可提高Ru-CeO2催化剂上生成苯的选择性, 这是由于Ru有较好的加氢和裂解H2O的催化活性所致。
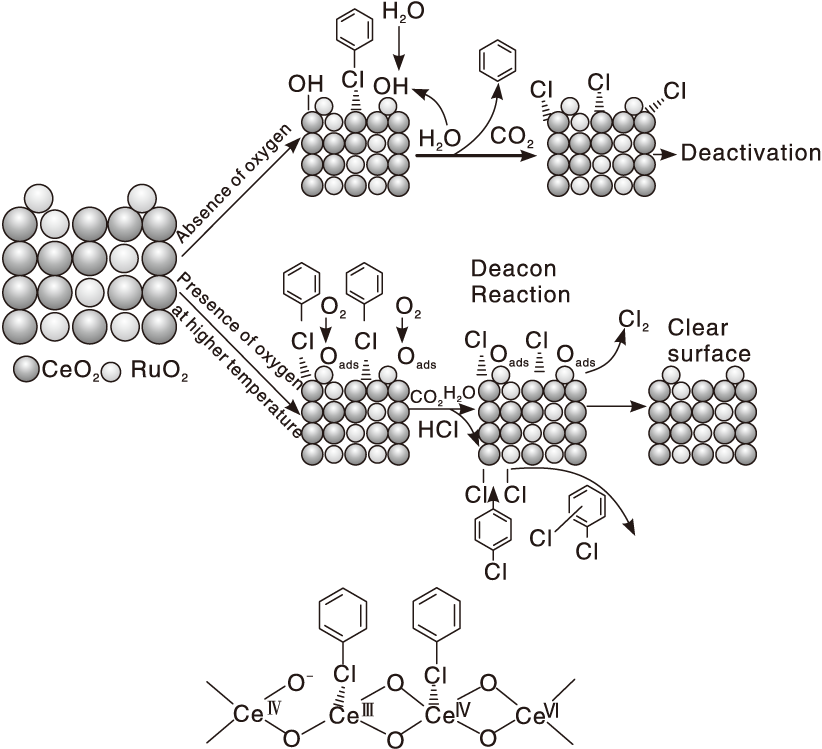 | 图5 氯苯在CeO2基催化剂上氧化反应机理[100]Figure 5 Proposed reaction mechanism for catalytic combustion of chlorobenzene over CeO2-based catalysts[100] |
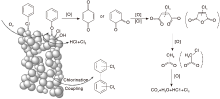
Liu X L等[101]利用原位红外光谱研究TiO2负载Pd、Pt、Ru、Rh等贵金属催化剂对氯苯氧化反应性能, 发现几种贵金属催化剂的反应中间产物相同, 由此推断其催化氧化反应机理也相似(图6)。氯苯先在活性位上解离吸附, 产生的酚盐物种进一步氧化生成邻位、对位苯醌, 随后在活性氧物种
对于CVOCs的催化氧化, 贵金属催化剂低温活性最好, 固体酸催化剂有丰富的表面酸中心, 而金属氧化物催化剂具有适中的酸性和氧化还原能力。因此, 金属氧化物催化剂近年来得到广泛深入的研究, 但其在较低温度下的抗中毒能力仍不够强, 催化反应机理尚有待于进一步探明。负载贵金属催化剂对CVOCs氧化的活性和CO2选择性较高, 缺点是产物中有较多的氯代副产物。由于催化剂的载体与贵金属之间存在强相互作用, 制备暴露高活性晶面的载体以及对载体进行掺杂改性是当前研究热点之一。在贵金属中, Ru具有最强的抗氯中毒能力和最低的氯化活性, 但金属态Ru易被氧化, 在CVOCs催化反应中Ru的配位环境对活性的影响尚不清楚。此外, 多元贵金属合金催化剂对CVOCs氧化反应鲜有报道, 也值得深入研究。
CVOCs分子在酸性位上吸附与活化、不同类型催化剂表面酸性的分布对CVOCs氧化活性和选择性具有一定的影响。L酸和B酸位对不同分子、基团的吸附强弱不同, 但并非所有酸性位均参与反应。CVOCs及其中间产物在催化剂的氧化还原位上进行深度氧化反应尚需进一步研究。对于饱和氯代烃的氧化, 提高催化剂的表面酸性相较于提高其氧化还原性能更有利于催化活性的改善, 而对不饱和氯代烃则相反, 这可能是由于不饱和氯代烃更难氧化以及在两种情形下反应的速控步骤不同所致。氧化还原位与酸性位之间存在协同作用, 如何建立催化剂的物化性质与催化性能之间的构效关系, 调控催化剂的元素配方、表面结构以及将这种协同作用最大化等课题还有待于深入研究。
在实际应用中, 污染源排放的CVOCs废气化学成分复杂, 水、碳氢化合物、氮氧化物等组分的存在会影响催化剂的性能。在模拟实际污染源排放CVOCs的条件下开展相应的研究工作, 将有利于研发更有实用价值的新型、高效、稳定的催化剂。由于催化剂的失活难以避免, 因此, 研发催化剂的再生技术也十分必要。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|