
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Co-N-C催化对甲基苯甲醇氨氧化合成对甲基苯腈
[吕庆阳, 夏语嫣, 马莉, 袁华*  ]
]
]
|
|


作者简介:吕庆阳,1993年生,男,在读硕士研究生。
以不同金属Co、Mn、Fe、Ni的醋酸盐为金属活性组分,1,10-邻菲啰啉为氮源,Vulcan XC72R型碳粉为载体,通过浸渍法负载后在不同温度下焙烧得到系列M-N-C催化剂。采用TEM、EDS、XRD、XPS、TG等对催化剂的结构进行表征分析,以对甲基苯甲醇氨氧化一步合成对甲基苯腈为探针反应考察M-N-C催化剂的催化氨氧化性能。结果表明,800 ℃焙烧的Co-N-C催化剂能够形成Co-N活性中心,具有较好的催化性能。在氧气分压0.5 MPa、反应时间24 h、氨水用量0.7 mL和反应温度130 ℃时,对甲基苯甲醇转化率94.4%,对甲基苯腈选择性96.4%。
A series of M-N-C catalysts were prepared by impregnation and calcined at different temperature with acetate of Co,Mn,Fe,Ni as active component source,1,10-phenanthroline as nitrogen source,and Vulcan XC72R type carbon powder as carrier.The prepared catalysts were characterized by TEM,EDS,XRD,XPS,TG,and tested in one-step synthesis of p-toluonitrile by ammoxidation of p-methylbenzyl alcohol.The results showes that Co-N-C catalyst calcined at 800 ℃ could form a Co-N active center and had good catalytic performance.When oxygen partial pressure was 0.5 MPa,reaction time was 24 h,the amount of ammonia was 0.7 mL and reaction temperature was 130 ℃,the conversion of p-methylbenzyl alcohol reached 94.4%,and the selectivity of p-methylbenzonitrile was 96.4%.
对甲基苯腈是广泛应用于医药、农药、荧光增白剂OB-1、KSN和DPP颜料等精细化学品的重要有机合成中间体, 经水解、还原还可分别转化为芳香羧酸或胺类等有机原料, 应用领域和市场需求不断增加。传统制备方法主要有以对甲苯胺为原料经过重氮化和Sandmeyer反应法[1]、以对甲基氯苯和NaCN为原料的Rosenmund-von Braun置换法[2]、甲苯催化氧化法[3]以及以醛、醛肟、酰胺、胺等为原料的合成法[4, 5, 6, 7], 但这些方法存在污染环境、工艺复杂、产率低等问题。
Mizuno N等使用氧化铝负载的氢氧化钌催化剂催化苯甲醇和氨水直接氧化合成对甲基苯腈, 收率达80%, 且副产物只有水[8], 为从醇直接氨氧化合成腈开辟了新途径。为避免使用贵金属, 采用非贵金属Cu、Fe等为活性组分直接催化对甲基苯甲醇氨氧化合成对甲基苯腈取得一定的进展[9, 10, 11], 但是与贵金属催化剂相比, 非贵金属催化剂催化活性明显降低, 因而使用均相的催化助剂提高催化活性。为了更好地适应当今绿色高效的生产需求, 需要寻求一种在非均相条件下由非贵金属催化合成对甲基苯腈的方法。
近年来, 氮掺杂碳材料中负载非贵金属形成的Mn-N-C催化剂在乙苯的氧化反应中显现出很好的催化活性[12], Co-N-C在醇选择性氧化为酮[13]和酯[14]的反应中表现出卓越的催化性能, Fe-N-C催化剂可高选择性氧化醇到醛[15], 表明在氧化体系中非贵金属M-N-C催化剂为可以与贵金属相媲美的非均相催化剂。本文选用不同非贵金属盐为金属活性组分, 以1, 10-邻菲罗啉为氮源, 形成不同的活性组分前驱体, 然后用浸渍法负载到Vulcan XC72R碳粉上, 通过焙烧法制备系列M-N-C催化剂。以对甲基苯甲醇直接氨氧化对甲基苯腈为探针反应, 探讨M-N-C催化剂催化性能, 为非贵金属非均相直接氨氧化对甲基苯甲醇的研究提供基础数据。
Co(OAc)2· 4H2O 、Cu(OAc)2· H2O、Mn(OAc)2· 4H2O、Ni(OAc)2· 4H2O、1, 10-邻菲啰啉、乙醇、叔戊醇、正十六烷, 阿拉丁试剂公司; O2、N2(99.9%), 武汉翔云化学气体有限公司; Vulcan XC72R碳粉, 美国卡博特公司。
称取0.475 g的Co(OAc)2· 4H2O、0.499 g的Cu(OAc)2· H2O、0.613 g的Mn(OAc)2· 4H2O和0.622 g的Ni(OAc)2· 4H2O分别至反应器中, 再加入0.901 g的1, 10-邻菲啰啉, 溶解于170 mL乙醇中, 室温搅拌30 min后, 再加入3.477 g的Vulcan XC72R碳粉, 60 ℃搅拌6 h。待混合液冷却至室温, 抽滤后置于70 ℃烘箱干燥过夜。在N2气氛焙烧2 h, 温度分别为400 ℃、600 ℃、800 ℃、1 000 ℃, 制得的催化剂标记为M-N-C/X(M为金属种类, X代表焙烧温度)。
TEM测试使用美国FEI公司Tecnai-G220-TWIN型透射电子显微镜。
X射线衍射能谱测试采用德国布鲁克公司QUANTAX系列能谱仪。
采用德国布鲁克AXS公司D8 ADVANCE X射线衍射仪进行XRD测试, 工作电压30 kV, 工作电流20 mA, 扫描范围5° 90° 。
采用VG Muililab 2000型X射线光电子能谱仪进行元素结合能分析, 所有结合能参比为C1S(284.8 eV)。
TG测试用美国TA公司Q50型热重分析仪, 升温速率10 ℃· min-1。
准确称取0.305 4 g对甲基苯甲醇于反应釜中, 加入0.2 g催化剂、适量氨水、20.0 mL叔戊醇, 密封, 通过进气装置向反应釜充入适量氧气, 设置相应的反应温度, 搅拌速率400 r· min-1, 反应一段时间后结束反应, 冷却至室温。打开反应釜, 加入0.1 g内标物正十六烷, 真空抽滤得滤液, 用50 mL乙酸乙酯稀释滤液。采用日本岛津公司GCMS-QP2010系列检测仪, 设置进样口温度为250 ℃, 柱箱温度从80 ℃以15 ℃· min-1的速率升温至280 ℃, 离子源温度220 ℃, 接口温度220 ℃。以正十六烷为内标物, 内标法计算对甲基苯腈收率和选择性。
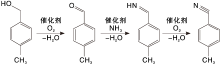
对甲基苯甲醇直接氨氧化合成对甲基苯腈的反应分为3步, 如图1所示。对甲基苯甲醇首先经选择性催化氧化得到中间体对甲基苯甲醛, 该步的关键是抑制醛的继续氧化; 对甲基苯甲醛再与氨加成消除反应脱去一分子水生成对甲基亚胺, 对甲基亚胺极不稳定, 迅速被氧化脱水得目的产物对甲基苯腈。
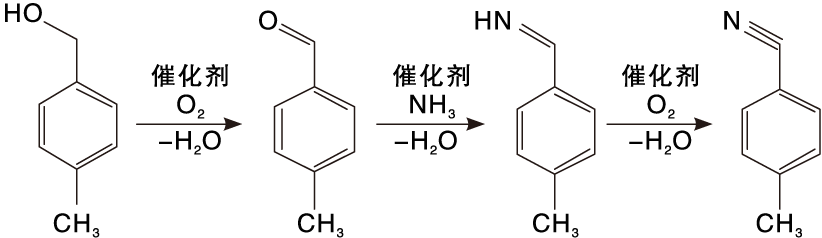 | 图1 对甲基苯腈的合成工艺路径Figure 1 Reaction path for the synthesis of p-methylbenzonitrile |
在反应温度110 ℃、反应时间20 h、氨水用量1 mL和氧气分压为1 MPa条件下, 不同金属组分得到的Co-N-C催化剂催化性能如表1所示。由表1可以看出, 焙烧温度为400 ℃时, 未负载金属组分的N-C/400基本没有催化活性; 加入金属组分以后, 对甲基苯腈收率提高, 其中Co-N-C/400催化剂上对甲基苯腈收率为28.54%, 提高幅度明显。
| 表1 金属盐组成对Co-N-C催化剂催化氧化对甲基苯甲醇性能的影响 Table 1 Catalytic oxidation of p-methylbenzyl alcohol over catalysts with different metal salt compositions |
对Co-N-C体系进一步优化, 改变Co(OAc)2· 4H2O与1, 10-邻菲啰啉物质的量比和焙烧温度, 结果如表2所示。
| 表2 金属盐组成与焙烧温度对Co-N-C催化剂催化氧化对甲基苯甲醇性能的影响 Table 2 Effects of metal salt compositions and calcinations temperature on catalytic performance of Co-N-C catalyst for oxidation of p-methylbenzyl alcohol |
由表2可以看出, 焙烧温度400 ℃、Co(OAc)2· 4H2O与1, 10-邻菲啰啉物质的量比为1∶ 2时, 对甲基苯腈选择性和收率最高; 焙烧温度800 ℃制备的Co-N-C/800催化剂上对甲基苯腈收率和选择性最高, 达到78.13%和94.32%。
2.3.1 TEM和EDS
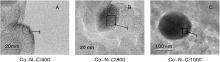
不同焙烧温度Co-N-C催化剂的TEM照片如图2所示。
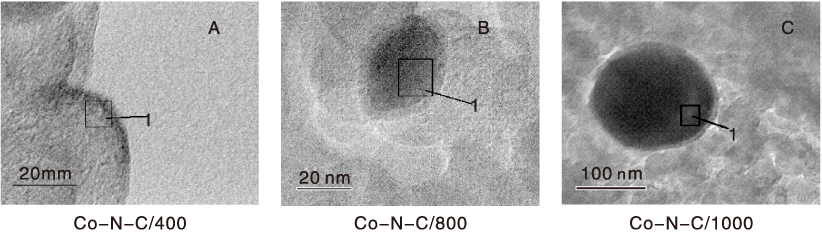 | 图2 不同焙烧温度Co-N-C催化剂的TEM照片Figure 2 TEM images of catalysts at different calcinations temperature |
由图2可以看出, 400 ℃ 焙烧的催化剂只有很小的团簇状微粒; 温度升高到800 ℃, 观察到直径(2030) nm的团聚物, 且包裹着一层(23) nm的物质; 温度进一步升高到1 000 ℃, 出现更大的团聚物, 直径超过100 nm, 外层被一层(68) nm的物质所包裹, 可能是随着焙烧温度升高, 金属组分烧结到一起的缘故。
选取不同焙烧温度Co-N-C催化剂中的特定区域(A-1、B-1和C-1)进行EDS能谱分析, A-1中Co质量分数为2.9%, B-1中Co质量分数为7.4%, 而C-1中Co质量分数高达24.0%, 结果如图3所示。图3证实了随着焙烧温度升高, 金属组分会出现团聚的现象。一般认为金属粒径越小, 分布越均匀, 催化剂催化活性越高。对比表12可知, 尽管Co-N-C/800的分散度不如Co-N-C/400催化剂, 但催化活性较高。
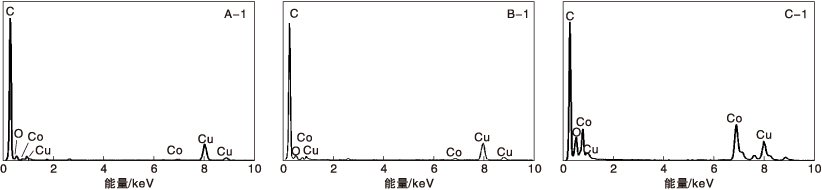 | 图3 不同焙烧温度Co-N-C催化剂的EDS能谱图Figure 3 EDS spectra of catalysts with different calcinations temperature |
2.3.2 XPS
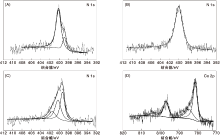
对不同焙烧温度Co-N-C催化剂进行全谱分析, 发现1 000 ℃焙烧后的催化剂检测不到N和Co元素, 结合TEM照片, 认为可能是因为1 000 ℃焙烧后的催化剂碳化严重, 碳层较厚, 而 XPS的常规分析深度为(510) nm。对未焙烧、400 ℃和800 ℃焙烧后催化剂N的结合能变化进行分析, 结果如图4(A、B、C)所示, 图4(D) 为Co-N-C/800催化剂的Co高分辨谱。
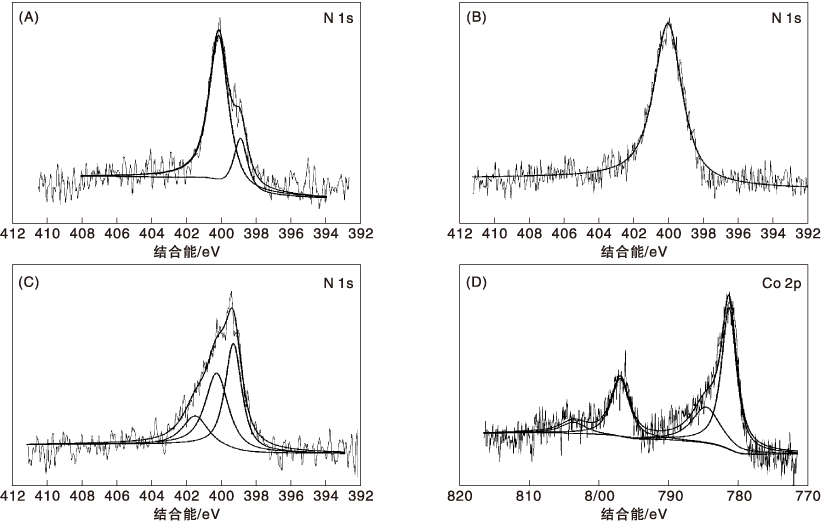 | 图4 不同焙烧温度Co-N-C催化剂的高分辨XPS谱图Figure 4 High resolution XPS spectra of catalysts at different calcinations temperature |
由图4(A、B、C)可以看出, Co-N-C/0催化剂的N高分辨谱图表明N1S可以分成两个峰, 结合能在398.92 eV和400.18 eV的峰分别归属于吡啶氮和吡咯氮; Co-N-C/400催化剂的N高分辨谱图表明N1s只有一个峰, 结合能400.02 eV的峰归属于吡咯氮, 吡啶氮的峰消失; Co-N-C/800催化剂的N高分辨谱图表明N1s可以分成3个峰, 结合能在399.14 eV、400.10 eV、401.33 eV的峰分别归属于Co-N中心、吡咯氮和石墨氮[16], 且Co-N中心对应的峰占主导地位。结合以上分析, 证明可能是Co-N中心的出现使催化剂催化活性得到显著提高。由图4(D) 可以看出, 780.90 eV和796.50 eV的峰归属于Co-N中心的2p3/2和2p1/2, 784.10 eV和803.10 eV的峰归属于Co-N中心的卫星峰[17]。没有检测到Co0或者Co3O4的特征峰, 与XRD结果吻合, 进一步证明可能是Co-N中心起着催化活性的作用。
2.3.3 XRD
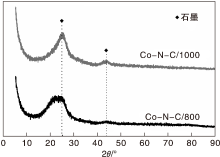
为了探究TEM图中团聚物是否为金属钴或者氧化钴, 对出现团聚的Co-N-C/800和Co-N-C/1000进行XRD表征, 结果如图5所示。
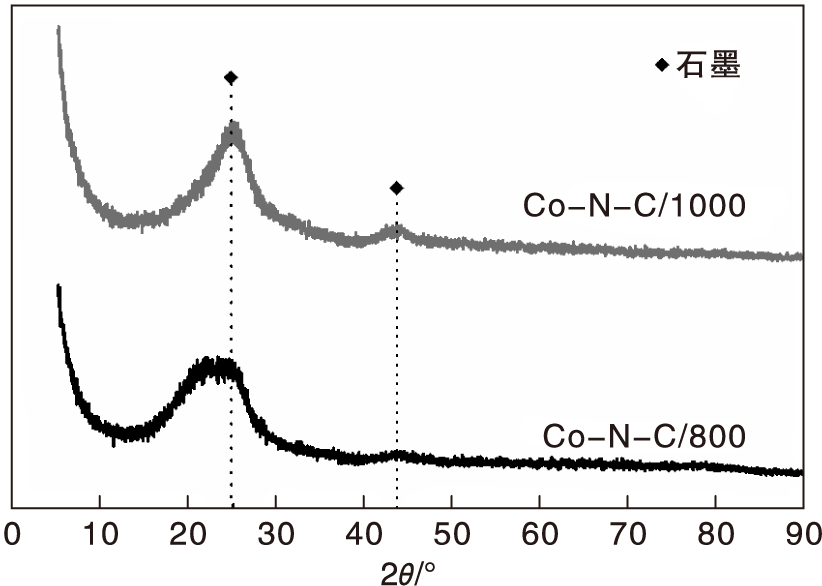 | 图5 Co-N-C/800和 Co-N-C/1000催化剂的XRD图Figure 5 XRD patterns of Co-N-C/800 and Co-N-C/1000 catalysts |
从图5可以看出, XRD图中只有C衍射峰, 且Co-N-C/1000的石墨化程度高于Co-N-C/800催化剂; 没有金属Co或Co3O4的特征峰, 表明Co可能以高度分散或无定型的形式存在。结合TEM照片, 活性最高的Co-N-C/800催化剂一方面分散度不如Co-N-C/400催化剂, 另一方面Co含量低于Co-N-C/1000催化剂, 因此, 可以认为催化剂活性中心并非是金属Co或Co3O4单因素所决定。
2.3.4 热重分析
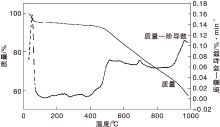
Co-N-C催化剂热重分析结果如图6所示。
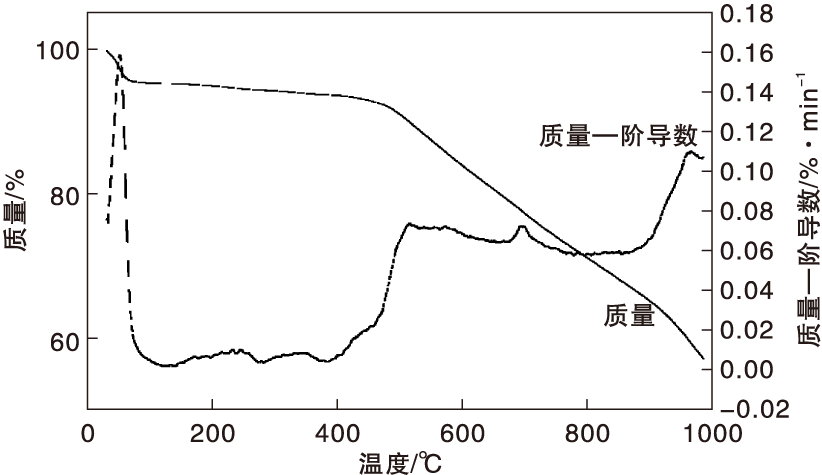 | 图6 Co-N-C催化剂热重分析曲线Figure 6 TG curves of Co-N-C catalyst |
由图6可以看出, 在约60 ℃, 催化剂有5%的质量损失, 是因为催化剂未干燥完全, 乙醇挥发所致; 在(100400) ℃, 催化剂质量较稳定; 高于400 ℃, 催化剂开始持续失重, 可能是1, 10-邻菲啰啉和醋酸钴分解所致; 在约700 ℃, 失重速率增强, Co-N-C活性结构形成; 900 ℃ 以后, 失重急聚加速, 催化剂碳化程度增强, 导致Co-N活性中心破坏, 催化性能急剧降低。
2.4.1 反应温度
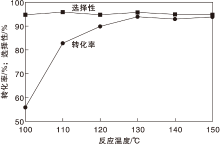
在氨水用量0.7 mL、反应时间24 h和氧气分压0.5 MPa条件下, 考察反应温度对Co-N-C/800催化剂催化性能的影响, 结果如图7所示。
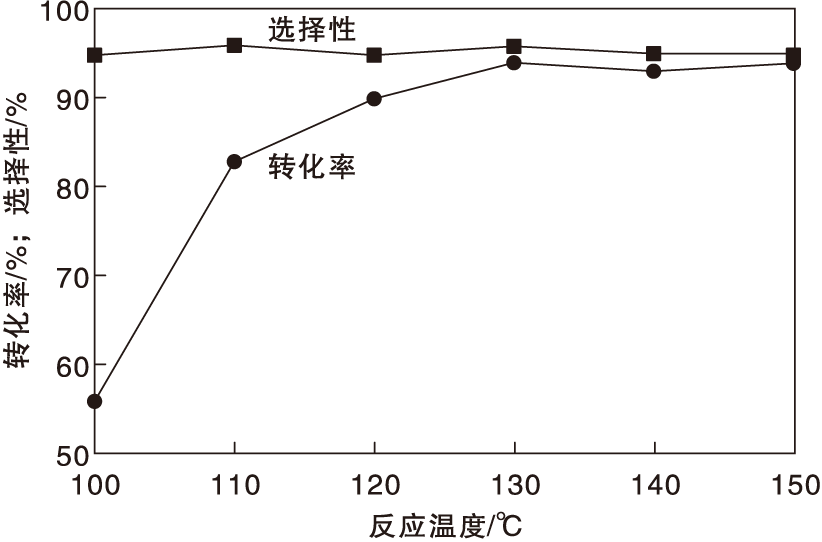 | 图7 反应温度对Co-N-C/800催化剂催化性能的影响Figure 7 Effect of reaction temperature on performance of Co-N-C/800 catalyst |
由图7可以看出, 反应温度从100 ℃上升到150 ℃, 对甲基苯甲醇转化率从56%提高到94%; 而反应温度对对甲基苯腈选择性影响不大。为减少能耗, 选择最佳反应温度为130 ℃。
2.4.2 氨水用量
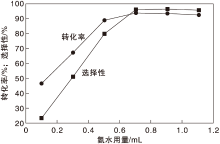
在反应温度130 ℃、反应时间24 h和氧气分压0.5 MPa条件下, 考察氨水用量对Co-N-C/800催化剂催化性能的影响, 结果如图8所示。
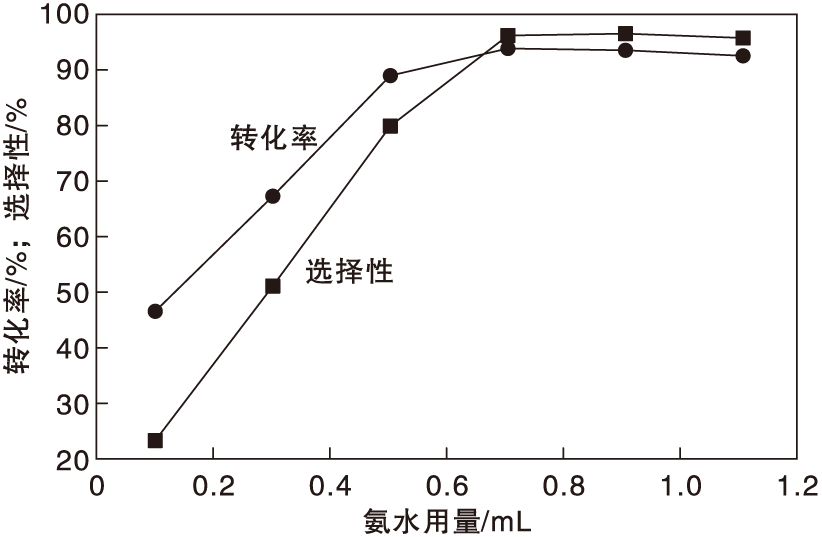 | 图8 氨水用量对Co-N-C/800催化剂催化性能的影响Figure 8 Effect of ammonia water amount on performance of Co-N-C/800 catalyst |
由图8可以看出, 随着氨水用量增加, 对甲基苯甲醇转化率和对甲基苯腈选择性随之增加, 当氨水用量为0.7 mL时, 反应达到平衡。继续增加氨水用量, 并没有其他副产物生成, 表明Co-N-C/800催化剂具有卓越的选择性。故实验选取氨水用量为0.7 mL。
2.4.3 反应时间
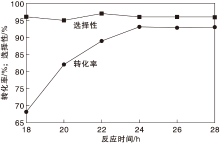
在反应温度130 ℃、氨水用量0.7 mL和氧气分压0.5 MPa条件下, 反应时间对Co-N-C/800催化剂催化性能的影响如图9所示。
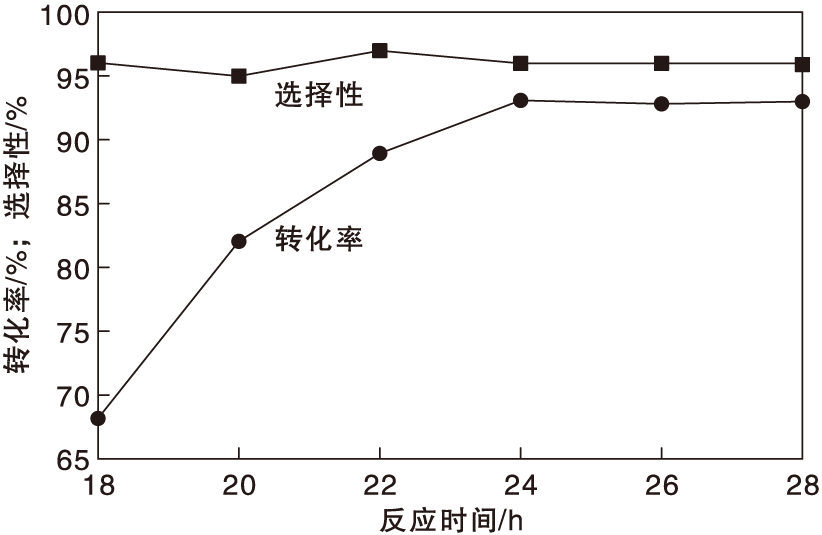 | 图9 反应时间对Co-N-C/800催化剂催化性能的影响Figure 9 Effect of reaction time on performance of Co-N-C/800 catalyst |
由图9可见, 反应时间从18 h增加到24 h, 对甲基苯甲醇转化率显著提高, 反应到24 h, 反应接近平衡, 对甲基苯甲醇转化率达到93.2%, 选择反应时间为24 h。
2.4.4 氧气分压
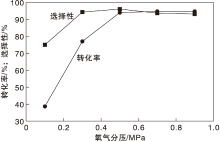
在反应温度130 ℃、氨水用量0.7 mL和反应时间24 h条件下, 考察氧气分压对Co-N-C/800催化剂催化性能的影响, 结果如图10所示。
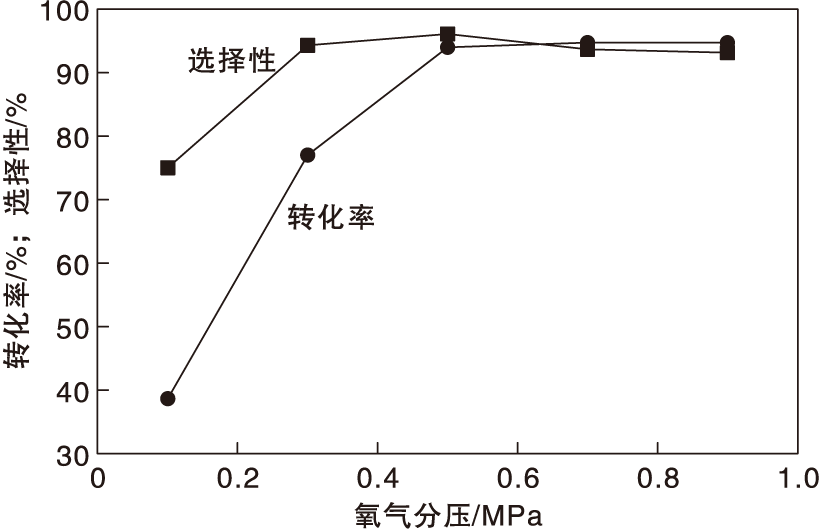 | 图10 氧气分压对Co-N-C/800催化剂催化性能的影响Figure 10 Effect of partial pressure of oxygen on performance of Co-N-C/800 catalyst |
由图10可见, 氧气分压为0.1 MPa时, 对甲基苯甲醇转化率38.3%, 对甲基苯腈选择性76.0%。随着氧气分压增加, 对甲基苯甲醇转化率和对甲基苯腈选择性提高; 当氧气分压为0.3 MPa时, 对甲基苯腈选择性提高到94.8%, 氧气分压达到0.5 MPa时, 对甲基苯甲醇转化率提高到94.4%, 对甲基苯腈选择性96.4%。继续增加氧气分压, 并没有过度氧化的现象, 表明催化剂具有优越的选择性。综合考虑, 选择氧气分压为0.5 MPa。
(1) 以对甲苯甲醇直接氨氧化制备对甲基苯腈为探针反应, 通过对不同金属组分和制备条件的筛选, 确定了以醋酸钴为金属前驱体、1, 10-邻菲啰啉为氮源、Vulcan XC72R碳粉为载体的催化体系, 在800 ℃焙烧得到的催化剂存在Co-N活性中心, 具有优越的催化性能。
(2) 在氧气分压为0.5 MPa、反应时间24 h 、氨水用量0.7 mL和反应温度130 ℃条件下, 对甲基苯甲醇转化率94.4%, 对甲基苯腈选择性96.4%。
致谢:感谢赵佳文、祝志巍、苏伟在测试工作中给予的帮助和指导!
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|