{kind=link}
{kind=link}
{kind=link}
催化裂化焦炭的生成及其对催化剂性能的影响
[于善青*  , 舒春溪, 李家兴, 林伟]
, 舒春溪, 李家兴, 林伟]
, 舒春溪, 李家兴, 林伟]
|
|


作者简介:于善青,1978年生,女,博士,高级工程师,主要从事催化裂化催化剂研发和海内外技术支持。
分析催化裂化过程中焦炭的生成及其对催化剂性能的影响,焦炭的生成与芳烃和烯烃等不饱和分子烃的环化、氢转移、烷基化以及缩合反应密切相关,在反应初期,焦炭产率处于较低水平且增幅不大,主要来自多环芳烃吸附焦;随着反应深度的增加,焦炭产率明显增加继而急剧增加,主要来自催化反应生成焦及对焦炭前驱物的吸附。焦炭对催化剂性能的影响主要表现在孔道堵塞和酸中心毒害,焦炭主要沉积在分子筛外表面或孔口处,使反应物分子不能进入孔内;残留在再生催化剂上的少量焦炭主要位于分子筛内,导致催化剂比表面积、孔体积和酸量的大量损失。基于以上分析,提出降低催化裂化焦炭的一些措施,为开发低焦炭产率的催化裂化催化剂和工艺提供依据。
The formation of coke and its effect on the performance of catalyst in FCC were analyzed.The formation of coke is closely related to the cyclization,hydrogen transfer,alkylation and condensation of unsaturated molecular hydrocarbons such as aromatics and olefins.At the initial stage of FCC process,the coke yield is at a low level and the increase is not significant,which is mainly from the adsorption coke of polycyclic aromatic hydrocarbons;With the increase of reaction depth,the coke yield increases obviously and then increases sharply,which mainly comes from the coke formed in catalytic reaction and the adsorption of coke precursors.The effect of coke on the performance of catalyst is mainly manifested in pore plugging and acid center poisoning.Coke is mainly deposited on the outer surface of the zeolites or at the pore openning,so that the reactant molecules cannot approach the acid site inside pore.A small amount of coke remained on regenerated catalyst is mainly located in zeolites,which leads to a large loss of specific surface area,pore volume and acid content of catalyst.Based on above analysis,some measures to reduce FCC coke are put forward,which can provide basis for developing FCC catalyst and process with low coke yield.
减少二氧化碳排放、缓解气候变化已经成为炼油行业转变经济增长方式、保持可持续发展的必由之路[1]。某炼厂对16套主要炼油装置的二氧化碳排放情况进行了估算[2], 其中催化裂化装置由于催化剂烧焦而成为二氧化碳排放大户。焦炭是催化裂化过程中的主要副产物, 催化剂在反应器中大量积炭后, 需要在再生器中烧焦再生, 焦炭燃烧后生成CO、CO2、H2O、SOx、NOx等, 燃烧产生的热量主要用于维持装置热平衡, 用于原料油的预热, 提供反应需要的热量, 给主风供热, 加热蒸汽等。但是随着催化裂化原料油不断重质化和劣质化, 焦炭产率增加, 烧焦过程产生的大量CO2所带来的环境问题更为突出, 并且导致热量过剩, 大量能源消耗。据统计, 催化剂烧焦再生过程能耗占催化裂化装置总能耗80%。因此, 降低催化裂化过程焦炭产率是实现炼油工业低碳化、降低能耗最有效手段之一[3, 4, 5]。
本文对催化裂化过程焦炭的生成及其对催化剂性能的影响进行分析探讨, 提出降低焦炭产率的一些措施, 为进一步开发低焦炭产率的裂化催化剂提供依据。
焦是一种不确定的含碳类沉积[6], 催化裂化催化剂焦炭的H/C物质的量比0.3~1.0。曹汉昌[7]提出一种根据烟气中CO2、CO、O2的体积分数测定催化裂化焦中氢含量的方法:
φ H/φ C=[8.93-0.425(φCO2+φ O2)-0.257φ CO]/(φ CO2+φCO)
根据此公式计算, 我国部分工业催化裂化装置焦中氢的平均质量分数为7.9%[8], 如果采用高活性分子筛催化剂, 在苛刻条件下焦中氢的平均质量分数为5%~6%。
按照焦炭的形成方式, 催化裂化过程主要生成4种类型的焦炭[9]:催化焦、附加焦、可汽提焦和污染焦。催化焦是烃类在催化剂中心通过催化裂化反应而生成的焦炭, H/C物质的量比约为0.4, 主要涉及催化裂化缩合和脱氢反应。催化焦的生成量与油剂接触时间的幂函数呈线性相关, 与剂油比成正比, 并随着反应转化率的增加而增加[10]。附加焦, 也称原料焦, 是原料油中生焦前躯物(尤其以胶质、沥青质等高沸点的稠环芳烃为主)在催化剂表面上发生吸附、缩合反应而生成的焦炭。附加焦与原料残炭值之间存在密切关系, 一般认为[8, 11]原料油的残炭有90%以上转化为附加焦, 与催化剂、反应时间和剂油比关联性较小[12]。可汽提焦, 也称剂油比焦, 是汽提不完全而残留在催化剂上的重质烃类, 并非真正的焦炭, H/C物质的量比较高, 属于富氢焦炭。可汽提焦与汽提段的汽提效率、催化剂孔结构等因素有关, 与反应时间无关[13]。污染焦主要来源于沉积在催化剂表面的重金属促进的脱氢和缩合反应产生的焦炭, 与沉积金属种类、金属沉积量以及催化剂抗金属污染能力有关。表1列出了催化裂化过程焦炭的类型、位置及影响因素[14]。
| 表1 催化裂化过程焦炭的类型、位置及影响因素 Table 1 Type, location and influencing factors of coke in FCC process |
随着催化原料的重质化, 催化裂化减压化中附加焦和污染焦导致大量焦炭的生成。表2为馏分油与掺渣馏分油催化裂化反应过程中4种焦炭分布和产率的比较[15]。由表2可以看出, 当减压馏分油中掺入一定量渣油后, 附加焦和污染焦的生成比例显著上升。
| 表2 馏分油与掺渣馏分油的催化裂化反应过程中焦炭的比较 Table 2 Comparison of coke in catalytic cracking reaction of distillate oil and residue blended distillate oil |
不同类型的焦炭对催化活性的影响不同, 与附加焦相比, 催化焦对催化活性的影响更显著。附加焦集中分布在催化剂外表面或基质大孔中, 容易毒害催化剂L酸中心; 催化焦主要分布在分子筛孔道中, 容易毒害催化剂B酸中心。所以相同焦炭含量下, 渣油生焦催化剂剩余活性高于轻油生焦催化剂[16]。就催化焦而言, 催化剂活性损失主要来自分子筛生焦失活, 分子筛生焦量大约是基质的10倍, 分子筛在1 s内活性损失90%, 基质大约需要1 min[14]。虽然基质的生焦影响较小, 但是基质在FCC反应中有促进反应物和产物分子传质、转热, 抑制分子筛生焦的作用。
按照焦炭的组成, 催化裂化焦炭可分为可溶性焦炭和不可溶性焦炭[17, 18, 19]。通常将待生催化剂先用有机溶剂处理, 洗去催化剂表面吸附的一部分积炭后, 用HF酸溶解分子筛骨架, 将不溶解的焦炭释放出来, 最后用有机溶剂(如CH2Cl2)处理上述释放出来的焦炭, 将溶于有机溶剂的组分归为可溶性焦炭, 将不溶组分归为不可溶性焦炭。研究表明[17]催化剂表面提取的物质主要是多环芳烃, 尤其是烷基取代的单环、双环、三环芳烃, 还含有少量的C9~C21烷烃, 它们对催化剂酸性和比表面的影响较小; 催化剂孔道内的可溶性焦炭主要是双元环芳烃至七元环芳烃; 催化剂上的不可溶性焦炭主要以无定形状态存在, 还含有一定量的高度缩合稠环芳烃。
催化裂化原料油包括饱和分、芳香分、胶质和沥青质。徐春明等[20]研究了胜利减压渣油的催化裂化反应特性, 认为生焦能力按照饱和分、芳香分和胶质顺序急剧增加, 这是由烃类本身的分子结构和生焦反应路径共同决定的。在催化裂化过程中, 烷烃和环烷烃主要以裂化反应生成小分子烯烃和烷烃为主; 芳烃主要发生侧链断裂反应和芳环缩合反应, 环数较小的芳烃可以断侧链减少碳数进入液体产物, 而环数较多的芳烃, 即使发生断侧链也很难进入汽油、柴油馏分段, 会进一步缩合生成焦炭, 可见芳烃对催化裂化生焦的作用是不言而喻的。崔维敏[21]探讨了减压蜡油中不同环数芳烃变化与焦炭的关系, 对于单环芳烃, 在反应初期(转化率不大于50%), 其物质的量产率增加不显著, 单环芳烃以芳烃迁移为主, 在反应中后期, 单环芳烃物质的量产率大幅增加, 表明有大量的非芳烃反应生成单环芳烃。对于双环芳烃, 在反应初期, 主要是芳环侧链的烷基和环烷基参与的裂化反应, 在反应中后期, 双环芳烃物质的量产率先升再降, 产率增加是由于非芳烃和单环芳烃经过环化氢转移等反应生成双环芳烃, 降低是因为随着反应深度的加深, 双环芳烃继续发生氢转移或环化脱氢缩合反应生成稠环芳烃甚至焦炭的能力增强。对于三环及多环芳烃, 在反应初期, 多环芳烃较多, 容易吸附在催化剂活性中心上进一步缩合生成更多碳数的芳烃甚至焦炭, 使其它烃类接触催化剂活性中心的概率降低, 影响其它烃类的裂化性能, 随着反应加深, 多环芳烃降低, 其它烃类接触活性中心的能力增强, 反应得到强化。
胶质中主要包括芳环部分、环烷环部分以及烷基侧链部分。在催化裂化过程中, 胶质主要发生烷基侧链的断裂反应和环烷环的部分开环裂化反应, 剩下的芳环部分可能直接进入液体产物中, 也可能继续在催化剂活性中心上发生缩合直至生焦。徐春明等[20]对胶质的催化裂化性能进行考察, 结果表明, 胶质的焦炭产率达到33%, 胶质裂化的汽油、柴油和气体产率之和达60%以上, 可见胶质的生焦能力较高, 但仍具有较好的裂化性能。
沥青质是不溶于正构烷烃(n-C5~n-C8)而溶于苯的一类物质, 是石油中相对分子量最大、极性最强的一种非烃化合物。研究[22, 23]认为劣质渣油沥青质分子中芳环数约10, 侧链总长度在C12以内, 以多取代、短侧链为主, 平均每个沥青质分子含有2个杂原子, 其中S以噻吩环为主, N以吡咯环为主。沥青质的生焦理论[24]认为沥青质胶粒进一步缔合形成的微观二维结构颗粒是成焦的基础, 二维结构颗粒可以形成宏观结构的焦炭, 其中二维结构颗粒的形成受沥青质分子之间的缔合作用影响, 沥青质分子由于π -π 键和氢键作用容易缔合, 进而会导致沥青质分子的絮凝、积垢、结焦。
催化裂化按照正碳离子机理进行, 主要包括烃类的裂化、氢转移、异构化和芳构化等。在催化裂化反应中, 最有可能发生的氢转移反应是1个环烷烃和3个烯烃反应生成1个芳烃和3个烷烃, 该反应被认为是生成焦炭的关键反应[25]。目前, 研究者普遍接受的反应机理如图1所示。由图1可知, 该反应主要可分为以下步骤:①环状碳正离子脱氢得到环烯烃; ②碳正离子和环烯发生氢转移反应; ③进一步失去氢质子, 得到环二烯并促进B酸的再生; ④环二烯和另一个碳正离子发生负氢离子转移; ⑤质子化的环二烯放出一个质子, 促进酸性位的再生, 同时产生一个芳烃分子。在上述过程中, 负氢离子转移为主要速率控制步骤。
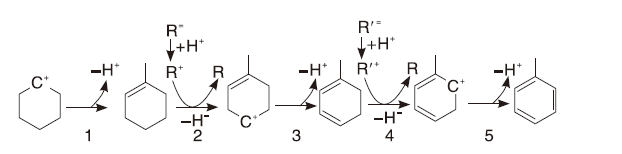 | 图1 氢转移机理示意图Figure 1 Schematic diagram of hydrogen transfer mechanism |
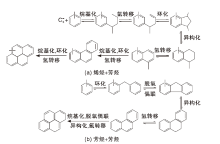
在常规催化裂化反应温度下(500 ℃~550 ℃), 第一个芳环的形成对于焦炭的生成尤为重要。在生成芳香分和烯烃等不饱和分子后, 生焦反应更容易进行。烯烃可以发生环化反应, 但同时也可以继续反应得到焦炭。而芳烃主要发生分子内的低聚和氢转移反应或是脱氢反应。因此, 催化裂化生焦可以分为两种不同的机理[26], 如图2所示。由图2可知, 芳烃和烯烃在B酸中心上进行烷基化反应, 生成的烷基化芳烃主要发生侧链氢转移和环化反应, 而后再经过异构化和氢转移反应生成萘类化合物。萘的衍生物通过相似的反应路径生成蒽、芘等化合物。芳烃+芳烃反应中, 主要是两个芳环发生烷基化反应后再发生脱氢耦合反应生成非芳香性的环戊环, 在异构化和氢转移反应后, 产生的蒽能继续发生烷基化、脱氢耦合、异构化和氢转移反应而生成芘, 甚至生成比芘更为复杂的化合物[27]。
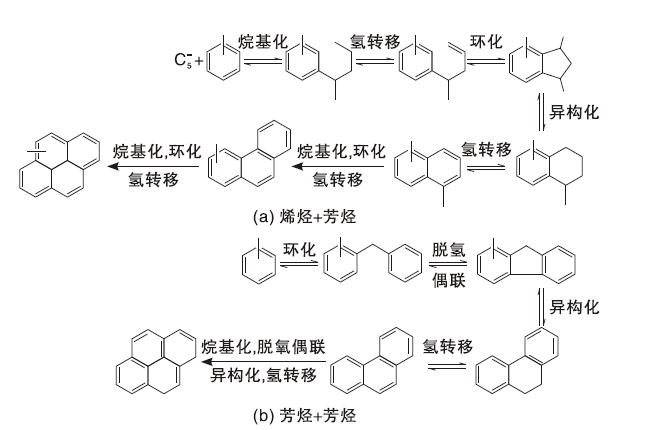 | 图2 焦炭形成机理示意图Figure 2 Schematic diagram of coke formation mechanism |
催化裂化生焦量受催化裂化转化率、反应时间、反应温度以及剂油比等因素的影响。
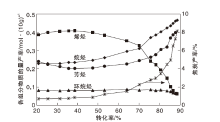
在不同转化率区间, 催化裂化焦炭的生成途径存在差异性[28, 29, 30, 31]。以镇海加氢蜡油催化裂化反应为例, 按照焦炭产率随转化率的变化趋势, 大体分为3个阶段[28], 如图3所示。
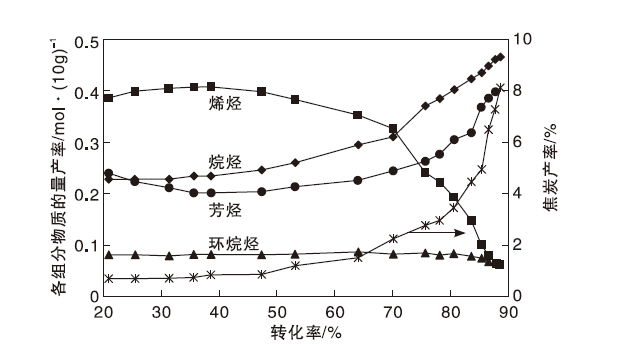 | 图3 镇海加氢蜡油催化裂化反应产物中汽油各组分物质的量产率和焦炭产率变化[32]Figure 3 Molar yield of gasoline components and coke mass yield in catalytic cracking of Zhenhai hydrogenated VGO[32] |
由图3可知, 第1阶段(转化率< 40%)为裂化反应开始阶段, 焦炭产率处于较低水平, 且随转化率的增加变化幅度不大, 焦炭主要来自多环芳烃吸附生成焦, 即原料中多环芳烃的吸附缩合反应。第2阶段(转化率40%~80%), 焦炭产率出现较大幅度增加, 这部分焦炭主要来自催化反应生成焦, 因为随着转化率的提高, 烯烃含量不断增加, 大量的烯烃分子不仅可以发生氢转移反应生成芳烃, 还可以继续发生氢转移反应生成稠环芳烃甚至焦炭, 烯烃与芳烃特别是多环芳烃的烷基化反应是焦炭产率升高的主要原因。第3阶段(转化率> 80%), 焦炭产率急剧增加, 此时催化剂孔道内已经吸附了较多的焦炭分子, 严重影响了气态分子在分子筛内部的扩散, 焦炭分子不断吸附焦炭前驱物、烯烃、芳烃以及其它焦炭分子, 缩合生成更大相对分子质量的焦炭, 使焦炭产率急剧增加[32]。
凌逸群等[33]研究了重油催化裂化提升管内反应时间对焦炭生成的影响, 将整个转化过程分为两个阶段, 即反应时间< 0.6 s油-剂初始混合段和反应时间(0.6~3.3) s油气渐次裂化段。在油-剂混合初期, 当原料油雾滴喷入提升管内与催化剂接触时, 较轻组分迅速气化, 而不能气化的重组分迅速润湿炽热的催化剂表面。这部分重组分中包含了原料残炭的主要“ 贡献者” , 这些“ 贡献者” 由于存在较多的“ 芳核” 结构, 分子尺寸较大, 不能有效扩散进入催化剂孔道进行裂化反应, 除了较长侧链发生断裂反应, 其中缩合程度较高的部分则在油-剂混合初期通过热缩合反应直接生成了附加焦。在油气渐次裂化阶段, 随着反应深度增加, 转化率增大, 更多的芳烃在提升管反应器后半段参加反应。
催化裂化焦炭产率随着反应温度升高而增加, 且增加趋势逐渐变得缓慢[34]。反应温度对焦炭产率的影响主要包括两个方面:1)随着反应温度增加, 低碳烯烃产率增加, 反应环境中烯烃浓度变高, 烯烃进一步聚合、芳构化生成焦炭; 2)焦炭也是缩合反应的产物, 而缩合反应是放热反应, 反应温度升高, 缩合反应减弱, 导致生焦量减少。此外, 催化裂化焦炭产率随着剂油比增加呈线性增加趋势, 剂油比增加意味着单位原料油接触催化剂活性中心的数量增加, 反应深度增加, 焦炭产率增加[34]。
在催化裂化操作中, 提高转化率的方式主要有两种, 即提高反应温度和增大剂油比。提高反应温度增加转化率, 焦炭选择性增加相对较少, 而干气选择性增加相对较多; 增大剂油比增加转化率, 干气选择性增加相对较少, 而焦炭选择性增加相对较多。在相同转化率条件下, 操作方式可以是较低再生温度和较高剂油比(方式一), 也可以是较高再生温度和较低剂油比(方式二), 如表3所示[35]。由表3可知, 当再生温度由720 ℃降低到610 ℃, 而剂油质量比由5.0增加到5.5 时, 干气质量分数下降0.29个百分点, 焦炭质量分数下降1.18个百分点。因此可以通过降低再生温度、增加剂油比来提高重油转化能力, 从而最小限度地增加非理想产物干气和焦炭的产率, 而最大程度地增加高附加值的液体产品。
| 表3 不同操作方式对产品分布的影响[35] Table 3 Effect of different operation modes on product distribution |
焦炭导致催化裂化催化剂失活的原因主要有两个:1)焦炭对催化剂孔道的堵塞; 2)焦炭对催化剂酸中心的毒害。
关于焦炭对催化剂孔道的堵塞, 一般认为[36, 37, 38], 焦炭沉积在分子筛外表面和孔口处, 降低了有效孔道尺寸, 阻碍了反应物分子与酸中心的可接近性。焦炭的沉积部位在很大程度上受分子筛孔道结构的影响[39], 对于中、小孔结构的分子筛(如ZSM-5), 结焦前驱体不能在孔内形成, 只有迁移到分子筛晶体外表面或接近外表面的孔口处, 才能进一步聚合为稠环芳烃至结焦; 用SiCl4消除HZSM-5外表面的酸性后与未处理的催化剂相比, 表面上形成的稠环芳烃比率下降, 推测最初在未处理的ZSM-5晶体内部形成的结焦前驱体, 可能在温度影响下迁移到外表面的酸中心上进一步缩合成稠环芳烃。对于大孔型或具有孔穴结构的分子筛[40, 41, 42](如HE、HY), 由于孔径大, 足以在孔道内形成稠环芳烃至焦炭并沉积下来, 同样是大孔的USHY、MOR和HERI分子筛的结焦速率并不相近, 其中USHY的结焦速率较其它两种分子筛的结焦速率更大, 因为USHY的孔径较大并且具有超笼结构, 有利于焦炭前驱体的扩散可以用“ 楔形理论” 解释分子筛孔道内的积炭[43], 把分子筛看成一个个圆柱体, 积炭则从靠近柱孔最外侧向内侧逐渐生成, 最外侧孔壁处积炭最厚, 最内侧只有很少积炭, 所以孔内活性损失并不是因为积炭直接覆盖在活性位上造成的, 而是因为积炭堵塞在孔口使反应物不能接近活性位, 从而使活性中心利用率大大下降。对于单维孔道结构的分子筛, 即使只有一个焦炭分子在孔道内, 也能阻碍反应物到达酸中心; 而具有超笼结构且具有三维孔道结构的Y分子筛的抗积炭性能比单维孔道结构的要好; 小孔径大孔穴的分子筛对焦炭失活也很敏感, 一旦小孔径被堵塞, 也阻碍了反应物到达活性中心[43]。
表4列出了两种催化剂的新鲜剂、待生剂和平衡剂的孔结构分析[17]。
| 表4 催化剂的比表面和孔结构分析 Table 4 Surface and pore structure of catalysts |
由表4可以看出, 与待生剂相比, 再生剂残炭量已经很低, 只有0.09%~0.13%, 但是比表面积和孔体积提高幅度不大, 只有5%~10%。可见, 再生剂上残留的少量积炭造成催化剂比表面积和孔体积大量损失, 也就是说再生剂上残留的这部分积炭是影响催化剂活性的主要因素。从保留度看, 待生剂的分子筛面积保留度远远低于基质面积保留度; 与待生剂相比, 再生剂的基质面积保留度提高幅度大于分子筛面积保留度的提高。进一步考察焦炭对催化剂微孔的影响, MLC-500新鲜剂微孔分布在0.786 nm和1.068 nm处; 积炭后, 0.786 nm处微孔消失, 1.068 nm处微孔移到1.141 nm, 吸附量显著降低; 再生后0.786 nm处微孔没有恢复, 1.068 nm处微孔吸附量仅提高了2.7%。以上结果表明, 分子筛提供的微孔受积炭影响很大, 微孔被积炭堵塞或者高温下被破坏, 再生也不能使微孔积炭完全去除, 也就解释了催化剂再生后表面积和孔体积的提高幅度不大的内在原因。
关于焦炭对催化剂酸性的影响, 普遍认为焦炭分子吸附在酸中心上, 线性影响催化剂活性, 进而影响反应选择性。对ZSM-5分子筛[44], 焦炭优先在强酸中心处生成, 尤其是强B酸中心, 再在弱酸中心上生成, 并且结焦速率随着酸度的增强而增大。Pierre Dejaifve等[39]考察了几种分子筛的结焦行为后认为, B酸中心在结焦中起主要作用, L酸对反应的一些步骤起到催化作用。表5列出了2种催化剂的新鲜剂、待生剂和平衡剂的酸性[17]。由表5可以看出, 与新鲜剂相比, 待生剂上B酸和L酸均有很大幅度降低, 表明焦炭覆盖了催化剂表面部分酸性中心或者堵塞了一部分微孔, 使催化剂酸中心减少; 根据Cerqueira H S[42]等的研究结果, 当积炭覆盖催化剂的酸中心时, 酸中心减少量与焦炭分子呈线性变化, 1个焦炭分子只能覆盖1个活性中心, 当积炭分子堵塞微孔时, 1个焦炭分子可以通过阻止反应物分子接近微孔内的酸中心, 从而使催化剂损失多个酸中心。B酸中心损失率远远大于L酸中心损失率, 焦炭更容易造成催化剂B酸中心的减少, 因为一方面B酸中心可以提供生焦反应的H+, 另一方面由于不饱和烃在强B酸中心上的吸附能力强, 不易脱附而聚集反应生成焦炭, 这是造成B酸损失大于L酸的原因。催化剂再生后, B酸和L酸只恢复了一小部分, 因为再生过程去除的是催化剂外表面或孔道外的积炭, 而外表面积炭对催化剂酸性影响不大, 真正影响催化剂酸性的积炭存在于微孔内或者孔口处, 而再生过程无法将这部分焦炭全部除去, 即使较少量的积炭也会造成酸性的大量损失。
| 表5 催化剂的酸性 Table 5 Acidity of catalyst |
在分析了催化裂化焦炭生成的相关机理以及其对催化剂性能的影响之后, 可以考虑采用以下方式降低催化裂化焦炭产率:
在炼油工艺方面:可以通过加氢处理和溶剂脱沥青等方法有效改善催化裂化原料油性质, 降低焦炭产率。例如, 溶剂脱沥青工艺可以有效脱除渣油中的沥青质及部分胶质, 由于渣油中所含的重金属绝大部分存在于胶质和沥青质中, 因此脱除沥青质及部分胶质的同时, 也可以脱除污染金属, 减少污染焦的形成[5]。还可以通过加入重油分散剂的措施来抑制沥青质胶体体系中胶粒之间的相互缔合和聚集, 从而有效改善重油体系的稳定性及反应性能, 防止生焦[27]。为了改善原料油的雾化性能, 可以借鉴重油乳化燃烧时的“ 微爆理论” 和“ 分子聚集和解散理论” , 在原料油中掺入水和乳化剂, 使之形成稳定的油包水型乳化液, 在高温下和催化剂接触时, 瞬间把包在外层的油相分裂成多个小油滴, 可以有效降低焦炭产率[45]。
在催化裂化操作方面:在油-剂混合初期, 优化油-剂接触温度, 减缓不能气化的重组分在催化剂表面通过热缩合反应生成附加焦的程度; 降低再生温度、增加剂油比来提高重油转化能力, 最小限度地增加非理想产物干气和焦炭的产率; 结合装置特点和原料油性质, 合理控制转化率, 过高转化率时焦炭产率急剧增加。
在催化裂化催化剂方面:添加抗重金属组元, 增加催化剂抗重金属污染能力, 尤其是捕捉Ni、V等污染金属的能力, 减少污染焦的形成; 具有良好的中大孔结构, 对于反应初期吸附在催化剂表面不能气化的重油大分子, 如果催化剂有较多的大孔, 可以增加这部分重油分子在催化剂上的扩散传质, 避免重油分子在某个活性位聚集生焦; 开发具有低氢转移活性和强裂化能力的分子筛组元, 氢转移反应对酸中心数量比较敏感, 而裂化反应对酸中心强度更加敏感, 因此适当降低Y 型分子筛的酸密度, 适当增加酸强度, 有利于降低氢转移反应同时提高裂化能力; 此外, 多级孔结构的分子筛有利于反应物的快速扩散, 也有利于降低氢转移反应几率。
(1) 催化裂化焦炭按照形成方式主要分为催化焦、附加焦、可汽提焦和污染焦, 不同类型焦炭在催化剂上的位置、影响因素及对催化活性的影响存在差异; 按照组成主要分为可溶性焦炭和不可溶性焦炭, 前者主要是双元环芳烃至七元环芳烃, 后者主要以无定形状态存在。
(2) 催化裂化焦炭的生成与不饱和烃的环化、氢转移、烷基化以及缩合反应密切相关, 其中第一个生成芳环的反应尤为重要, 在生成了芳香分和烯烃等不饱和分子后, 生焦反应更容易进行。在催化裂化反应初期, 焦炭产率处于较低水平并且增加幅度不大, 焦炭主要来自多环芳烃吸附焦; 随着反应深度的增加, 焦炭产率明显增加继而急剧增加, 焦炭主要来自催化反应生成焦及后期对焦炭前驱物的吸附。
(3) 催化裂化焦炭对催化剂性能的影响主要表现在对催化剂孔道的堵塞和对催化剂酸中心的毒害, 焦炭主要沉积在分子筛外表面或孔口处, 进而使反应物分子不能接近孔内酸性中心; 焦炭的沉积导致催化剂酸中心的明显降低, 相比于L酸中心, B酸中心损失更显著。催化剂再生过程无法完全除去分子筛内的焦炭, 即使少量焦炭也导致催化剂比表面积、孔体积和酸量的大量损失。
(4) 结合炼油工艺、催化裂化操作和催化剂研发, 提出了降低催化裂化焦炭的一些措施, 以期为炼油工业低碳化和低能耗的发展提供理论依据。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|