{kind=link}
{kind=link}
NO催化氧化催化剂研究进展
[杨玲, 王晗, 马幸, 郭锋, 梁婉*  ]
]
]
|
|


作者简介:杨 玲,1986年生,女,上海市人,博士,从事烟气脱硫脱硝催化剂研究。
氮氧化物(NOx),尤其是一氧化氮(NO)是最主要的空气污染物之一,可导致光化学烟雾、酸雨等环境问题。NO催化氧化成二氧化氮(NO2)是消除NOx的关键所在。近几十年,NO氧化催化剂研究广泛,主要分为4类:负载型(包括贵金属和金属氧化物),多元金属氧化物,钙钛矿以及碳基催化剂。主要概述NO氧化催化剂的研究进展,催化性能以及反应机理和抗SO2/H2O性能。最后对NO氧化催化剂的发展前景和方向进行展望。
Nitric oxides (NOx),especially NO,are one of the major air pollutants,which cause environmental problems,such as photo-chemicalsmog,acid rain and so on.The catalytic oxidation of NO to NO2 is the key step in NOx elimination.In recent decades,NO oxidation catalysts have been widely studied.The catalysts were divided into four series,including supported noble metal and metal oxides catalysts,multi-metal oxides catalysts,perovskite type catalysts and carbon-based catalysts.This paper generally summarized the research progress on catalysts,evaluation of catalytic performance in NO oxidation,mechanistic investigations and SO2/H2O tolerance.Finally,the perspective and the future direction of NO oxidation catalysts were presented.
氮氧化物(NOx, NO、NO2、N2O等)排放导致一系列环境问题, 如光化学烟雾、酸雨等, 且NOx排放可加速形成二次气溶胶和细颗粒。因此, 人们对如何降低NOx的排放做了很多的研究, 并提出了非常多的途径, 如NOx储存还原(NSR), 三效催化剂, 连续再生颗粒捕集(CRT), 以及选择性催化还原(SCR)等[1, 2, 3, 4, 5]。在NSR中, 通过贵金属将NO氧化为NO2, 再以硝酸盐或亚硝酸盐的形式储存在碱性组分上[6, 7]。在CRT碳烟颗粒的去除中, NO同样先氧化成NO2, 再将柴油机颗粒过滤器收集的碳烟颗粒氧化[8]。
NH3-SCR被认为是最有效的降低NOx排放的技术[9, 10, 11, 12]。NO部分氧化成NO2后, NH3-SCR反应速率大大提高, 快速SCR反应(4NH3+2NO+2NO2=4N2+6H2O)速率在低温[(200~300) ℃]时是NO和NH3基础反应(4NH3+4NO+O2=4N2+6H2O)的10倍[13, 14, 15]。NO氧化成NO2是NH3-SCR反应中非常重要的一步。然而NO的氧化效率在标准条件下较为缓慢, 这主要是由于气相中化合物的化学性质比较复杂, 以及不同的颗粒物分布。研究合适的NO氧化催化剂得到了非常多的关注。
本文着重介绍NO氧化催化剂的研究进展。催化剂主要分为负载型催化剂、多元金属氧化物催化剂、钙钛矿型催化剂、活性炭催化剂等, 总结了不同催化剂对于NO氧化的催化活性, 反应条件, 以及反应机理研究和SO2/H2O中毒影响。
负载型催化剂中活性组分主要存在于催化剂的表面, 载体提供了巨大的比表面积, 不仅可以分散活性中心, 同时也提供了催化反应的空间。
1.1.1 负载型贵金属催化剂
(1) 单一贵金属催化剂
Pt催化剂是最早用于NO氧化的单一贵金属催化剂[8, 16, 17]。载体种类, Pt负载量, Pt分散度, Pt氧化物的形成方式等均影响Pt催化剂在NO氧化反应中的活性[18, 19, 20]。Schmitz P J等[21]研究了负载型Pt催化剂对NO氧化的影响, 发现载体的影响> 预处理> 负载> 焙烧气氛> 焙烧温度> 前驱体盐。Xue E等[18]发现Pt/SiO2活性> Pt/γ -Al2O3> Pt/ZrO2。负载量为质量分数0.1%~5%时, Pt催化剂活性随负载量的增加而提升, Pt/SiO2催化剂中Pt颗粒越大, 活性越高, 表明催化剂具有明显的尺寸效应。Olsson L等[19]证实NO氧化的反应速率随Pt颗粒尺寸的减小而降低, 这主要是由于小颗粒Pt易形成PtO和PtO2, 氧化态Pt的催化活性低于金属态。
在多相催化反应中, TiO2也是一种优异的催化剂载体[22, 23]。Li Landong等[8]采用湿法浸渍(imp)和光沉积法(pho)制备了Pt/TiO2催化剂。结果表明, 湿法浸渍Pt/TiO2催化剂用H2/He预处理比O2/He预处理活性更高, 光沉积制备的Pt/TiO2催化剂O2/He预处理后表现出最好的NO氧化活性, 在反应温度为450 ℃时, NO转化率可达到90%以上。更深入的研究表明, 湿法浸渍制备的Pt/TiO2催化剂O2/He预处理后主要包含PtⅡ O, 这种形态比金属态Pt的活性低得多[19, 24]。光沉积Pt/TiO2催化剂中, 不同条件预处理后, Pt的形态通常是Pt0, Pt分散性比湿法浸渍催化剂好, 且催化活性也更高。Song Shuzhen等[25]发现, 沉积-沉淀法制备Pt/TiO2催化剂过程中, pH值影响催化活性, pH值约为7时, 催化剂具有最高的活性。
Li Landong等[26]制备了一系列Ru催化剂, 包括Ru/TiO2(P25, 70%锐钛矿, 30%金红石)、Ru/TiO2(锐钛矿, A)、Ru/TiO2(金红石, R)、Ru/ZrO2、Ru/SiO2和Ru/Al2O3, 并用于NO氧化反应。结果表明, Ru/TiO2催化剂活性最好, 在反应温度为275 ℃时, NO转化率可达到94%。混合相TiO2载体比单一相TiO2效果更好, 当Ru负载量为质量分数2%时活性最佳。研究发现, 在Ru催化剂表面形成了不同种类的RuO2物种, 其中高度分散的无定型RuO2的活性最高。Pd也是一种典型的贵金属催化剂, 但是Auvray X等[27]发现Pd/Al2O3催化剂活性低于Pt/Al2O3和Pt-Pd/Al2O3, Narula C K等[28]也证实Pd/Al2O3催化剂即使在反应温度354 ℃时也仅有17%的NO转化率。在Al2O3(010)面的Pd单原子对于NO氧化没有活性, 因为在催化剂上没有硝酸盐的形成, 另外, NO2的脱附需要能垒, 这阻碍了NO氧化。Weiss B M等[29]发现NO氧化转化速率随着PdO颗粒的减小而降低, 这主要是由于小颗粒中的氧空位较少, 氧的键能较大, 而且氧的活化是控速步骤。除了Pt、Ru和Pd, Au纳米颗粒也是NO氧化潜在的光催化剂, Herná ndez-Ferná ndez J等[30]发现Au负载量为质量分数0.5%时, Au-TiO2光催化剂具有卓越的催化活性, 在紫外光下, 60 min内, NO转化率可达到85%。Zhang Dieqing等[31]研究了Au纳米颗粒均匀分散在金红石型TiO2纳米棒上的催化剂, 结果表明, 在光催化NO氧化反应中, 该催化剂具有高活性, 寿命长的特点, 原因主要是Au量子点和TiO2纳米棒间的相互促进作用可产生光敏性, 活化NO分子, 减少光电子穴重组, 稳定Au量子点。
(2) 掺杂型贵金属催化剂
Dawody J等[32]研究金属掺杂对Pt/Al2O3催化剂的作用, 掺杂金属包括WO3、MoO3、V2O5、Ga2O3。结果表明, WO3/Pt/Al2O3具有最高的NO氧化活性, MoO3/Pt/Al2O3次之, 且添加MoO3减小了SO2对反应的影响。Irfan M F等[33]发现WO3可以抑制Pt的氧化从而提高NO氧化的催化活性。Hauff K等[34, 35]指出WO3和MoO3金属氧化物的加入可增强催化剂的氧化还原性能。Liu Shuang等[36]发现MnOx-CeO2混合氧化物掺杂Pt/Al2O3催化剂也可以提升催化活性, 在反应温度350 ℃时, NO转化率可达到80%。Zhang Hailong等[37]分别将Al2O3、ZrO2、La2O3和Y2O3引入Pt/CeO2-MnOx催化剂中提高热稳定性, 经过热处理后, NO氧化活性依然较好。
(3) 双金属Pt-Pd催化剂
Kaneeda M等[38]证实了在Pt/Al2O3催化剂中加入Pd可以阻止Pt物种的烧结, 提高热稳定性, 但是Pd加入降低TOF值, 这主要是由于Pd没有Pt的活性高, Pd的最大添加量为0.3%(物质的量分数)。Auvray X等[27]采用共浸渍和分步浸渍法合成Pt-Pd/Al2O3催化剂, 发现分步浸渍法合成的Pt-Pd/Al2O3催化剂具有最高的活性, 反应温度为250 ℃时, 活性可达到91%。若反应气氛中存在碳氢化合物, 如丙烯等, 会降低Pt-Pd/Al2O3对NO氧化的活性, 主要是由于Pd的加入在很大程度加速丙烯的氧化, 阻碍NO氧化[39, 40]。
1.1.2 非贵金属氧化物催化剂
尽管贵金属催化剂具有卓越的NO氧化性能, 但是较高的价格限制了其应用, 而且其失活原因仍然不是很清楚。负载型金属氧化物催化剂因低成本、高活性和稳定性得到了广泛研究。
(1) Al2O3负载金属氧化物催化剂
Al2O3具有高热稳定性、较大比表面积、较多酸中心, 易于NO物种的吸附[41, 42]。Nickolov R等[43]发现Mn2O3改性γ -Al2O3载体负载CuO可以阻止CuO的聚集以增强催化剂活性。Wang Pan等[41]通过溶胶凝胶法合成了一系列xMn10Ce/γ -Al2O3催化剂(x=4、6、8、10), 催化剂具有较好的结晶性和分散性, 其中6Mn10Ce/γ -Al2O3催化剂显示出最好的NO氧化活性, 在反应温度为300 ℃时, 转化率可达到83.5%。
(2) TiO2负载金属氧化物催化剂
TiO2的酸中心虽然没有Al2O3多, 但其具有更好的分散性, 由于硫酸盐易于在TiO2表面分解[44], 所以抗SO2性能较佳。Wu Zhongbiao等[45]用沉积沉淀法制备了一系列不同Mn/Ti比例的MnOx/TiO2催化剂, 其中Mn/Ti物质的量比为0.3时, 活性最好, 在反应温度为250 ℃时, NO转化率可达到89%, 表征结果显示, 在此比例下MnOx在TiO2表面具有非常好的分散性, 以及Mn3+物种的存在。An Zhongyi等[46]研究了TiO2晶相(金红石、锐钛矿、P25)对MnOx/TiO2催化剂的影响。结果表明, 含10% P25相催化剂活性> 锐钛矿相> 金红石相, 原因是P25具有更好的分散性, 降低活性颗粒的团聚。Li Xiaohai等[44]研究Ce掺杂对MnOx/TiO2催化剂的促进作用。结果表明, Ce的引入增加了催化剂的储氧能力以及氧的流动性。Zhang Jingxin等[47]合成了一系列不同Cr/Ce比的Ce掺杂CrOx-TiO2催化剂。结果表明, Cr(1)Ce(0.25)/TiO2催化剂性能最佳, 在反应温度为350 ℃时, NO转化率为69%。表明适当添加Ce可以增强金属与载体之间的相互作用, 增加还原相, 更易于反应的进行。
(3) ZrO2负载金属氧化物催化剂
ZrO2对NOx具有较高的吸附能力, 对NO氧化有潜在的应用[48, 49, 50]。Zhao Baohuai等[51]研究了MnOx/ZrO2催化剂。结果表明, 与MnOx/TiO2相比较, 该催化活性和热稳定性均更好。ZrO2除了作为载体, 还为硝酸盐中间物种提供了大量的吸附位, 更易于NO氧化。Zhao Baohuai等[52]通过柠檬酸络合法一步合成了Co/Zr1-x-CexO2催化剂, 在反应温度为300 ℃时, NO转化率可达到79.8%。
不同组成的多元金属氧化物的协同作用可以提高催化剂活性, 虽然多元金属氧化物催化剂已被广泛研究, 但相比较而言, Mn-Ce-Ox和Ce-Co-Ox催化剂在低温下对NO氧化催化活性最高。Mn物种具有多种价态, 在多元金属氧化物催化剂中表现出优异的活性[52, 53]。CeO2由于其良好的储氧性能和氧化还原性质得到广泛研究[54, 55, 56]。Sun Yuanyuan等[57]研究了稀土金属(Ce、La、Pr)掺杂对Mn基催化剂NO氧化的促进作用。在反应温度为239 ℃时, NO氧化转化率最高可达94%。Wang Zhihua等[58]研究了Co、Mn、Fe、Cr、Ni修饰CeOx催化剂的NO氧化性能。结果表明, Co修饰的催化剂具有最高的催化活性, 在反应温度为230 ℃时, NO转化率可达到90%以上。Ce-Co催化剂优异的活性与其结构、结晶性、氧化还原性, 以及Co与Ce之间的相互作用有关。Li Hua等[59]报道Mn-Ce-Ox催化剂在反应温度(100~200) ℃具有优异的催化活性, 在反应温度为150 ℃时, NO转化率即可达到60%以上。Ce的引入不仅可以增加MnOx的比表面积, 还可以提高Mn物种的分散度。Cui Mingshan等[60]研究了共沉淀法制备的MnOx-CeO2催化剂。结果表明, MnOx与CeO2之间较强的相互作用增强了氧化物的还原能力, 以及Mn4+的含量。Li Kai等[61]发现Mn-Co-Ce-Ox复合氧化物在较低温度下具有较高的活性, 在反应温度为150 ℃, 空速GHSV=35 000 h-1时, NO转化率可达到80%以上。与Mn-Co-Ox相比较, 催化剂的比表面积、孔容、孔径都有明显的提高。Wang Zhong等[62]研究了Cu/Ce0.8Zr0.2O2催化剂对NO氧化的性能, 适当添加Cu后, 催化性能提升, 同时具有一定的抗H2O和SO2性, 载体表面CuO物种的分散性对NO的吸附有一定的影响, 而且Cu-Ce-Zr固溶体提供了氧空位形成活性氧, 提高催化剂NO氧化的活性。
近年来, 钙钛矿型氧化物已成为最具潜力的催化剂, 其成本低、活性高、热稳定性好, 这类材料的通式为ABO3, A位是稀土金属或碱金属离子, B位是过渡金属, 通过A或B位与其它离子的部分调节, 即可改变氧化还原性能[63, 64, 65]。LaMnO3或LaCoO3钙钛矿A位取代明显提升活性[66, 67]。Chen Jiahao等[68]通过溶胶凝胶法合成了非计量比的LaxMnO3(x=0.9、0.95、1、1.05、1.11), La0.9MnO3表现出最高的活性, A位低含量的La促使Mn3+向Mn4+转变, 以平衡价态和结构稳定性。Shen Meiqing等[69]研究了Ca取代LaMnO3对NO催化氧化的作用, La0.9Ca0.1MnO3具有优异的NO氧化性能, 在反应温度为300 ℃时, NO转化率为82%。Ca部分取代后, 增加了Mn3+及表面吸附氧的数量, 促进NO氧化。Yoon D Y等[70]通过溶胶凝胶法合成Ag掺杂钙钛矿催化剂La1-xAgxMnO3, 其中La0.8Ag0.2MnO3催化剂性能最好, 主要是由于其具有较高的氧空位。
钙钛矿B位部分取代同样影响催化性能。Chen Jiahao等[71]研究了溶胶-凝胶法制备的LaMeO3(Me=Mn、Fe、Co)催化剂, 发现LaCoO3比其它两种催化剂活性高, 这主要是由于催化剂的还原性能不同。Wen Yuxin等[72]研究Ce掺杂钙钛矿催化剂(L
活性炭和活性炭纤维催化剂具有优异的吸附性, 氧化还原性, 以及较多的酸中心[74, 75, 76, 77], 是NO氧化的良好催化剂之一。Guo Zhanchen等[78]研究了椰壳活性炭、沥青基活性炭纤维和聚丙烯腈基活性炭纤维催化剂。结果表明, NO转化为NO2的平衡状态主要取决于温度、O2浓度、以及活性炭性质。催化活性顺序为聚丙烯腈基活性炭纤维< 沥青基活性炭纤维< 椰壳活性炭, 其中椰壳活性炭在低浓度O2条件下依然表现出较高的NO氧化活性。Adapa S等[79]研究了NO在活性炭纤维上催化氧化的影响因素。结果表明, NO转化程度取决于反应温度、O2浓度和活性炭纤维的种类。与沥青基和胶黏人造丝基活性炭纤维相比较, 酚醛树脂基活性炭纤维对NO氧化更有效。Mochida I等[80]发现湿度对沥青基活性炭纤维活性具有抑制作用, 在干燥空气中, 室温下NO转化率达到87%, 在相对湿度80%的空气中, 转化率降至62%, NO的吸附受湿度的影响。
近年来, N掺杂碳材料的研究越来越多[81, 82]。碳材料表面N的存在增强吸附酸性气体的能力[83], 对催化活性的影响主要是提供表面碱性位和提供电子[84], 含N表面基团的存在也对NO氧化成NO2非常有帮助。Sousa J P S等[1]制备了一系列具有不同N组分的活性炭, 其中采用尿素水溶液处理的活性炭催化活性最好。该研究者还发现硝酸以及三聚氰胺处理的活性炭也对NO氧化有积极作用[85]。Rathore R S等[86]将活性炭纤维采用氨气功能化, 用于碳烟颗粒去除, NO氧化转化率70%左右, 这主要是由于氨功能化活性炭纤维表面具有含N基团, 易于NO的吸附, 并且室温下比高温下效果更好, 该催化剂也可以捕获颗粒物质, 颗粒物质减少约90%。
催化剂的动力学主要取决于以下几个方面:(1)反应物的吸附; (2)催化反应的活性位点; (3)反应产物的脱附。NO在催化剂表面的氧化主要有两种反应机理:(1)Eley-Rideal(E-R)机理:NO(O2)先吸附在Lewis酸或Brø nsted酸位, 然后吸附态的NO(O2)直接与气相O2(NO)反应生成吸附态NO2; (2)Langmuir-Hinshelwood(L-H)机理:NO和O2同时吸附在催化剂的活性位上, 然后NO氧化成NO2。
Olsson L等[87]建立了Pt/Al2O3催化剂上NO氧化的E-R模型, Pt-O+NO→ Pt-NO2被认为是决速步骤。Xue E等[18]发现Pt催化剂上NO氧化中, Pt和载体的键能是非常重要的因子。Li Landong等[88]认为采用Pt/TiO2作为催化剂时, 当温度低于表面硝酸盐分解温度时, 气相NO直接氧化是形成NO2的唯一路径。表面硝酸盐物种的分解在NO氧化中起到明显的反作用, 原因是其分解产物主要是NO, 而不是NO2。
此外, Bhatia D等[89]研究了Pt/Al2O3上NO氧化动力学, 根据L-H机理提出不同的NO氧化路径, O2吸附被认为是决速步骤, 反应机理如下所示:
NO+Pt→ NO-Pt
O2+2Pt→ 2O-Pt
NO-Pt+O-Pt→ NO2-Pt+Pt
Pt-NO2→ NO2+Pt
Adapa S等[79]提出了在使用ACF作为催化剂时, NO氧化同时遵循L-H机理和E-R机理, 如表1所示, O2有两种可能的状态。
| 表1 ACF催化剂上L-H机理和E-R机理 Table 1 L-H mechanism and E-R mechanism over ACF catalysts |
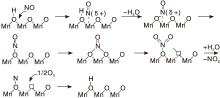
Tang Nian等[90]研究了MnOx/TiO2催化剂上NO催化氧化的机理, 最可能的过程如图1所示, NO先吸附在金属位, 形成亚硝酰基, 易于形成硝酸盐, 然后硝酸盐分解, 产生NO2并脱附。
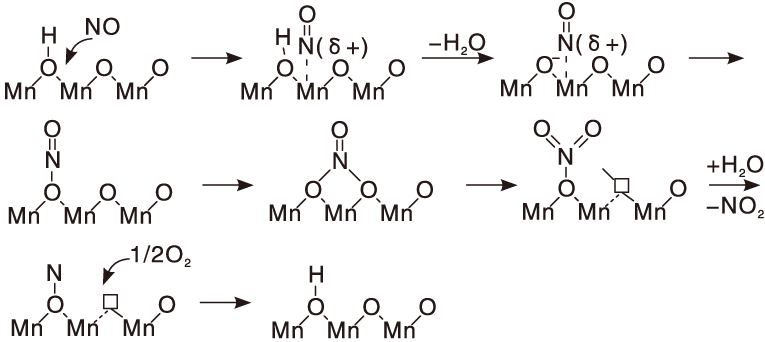 | 图1 MnOx/TiO2催化剂上NO氧化主要路径Figure 1 Main pathway of NO oxidation over the MnOx/TiO2catalyst |
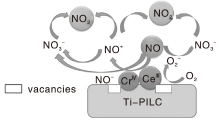
Zhong Lei等[91]提出了CeO2改性Cr/Ti-PILC催化剂上NO氧化可能的反应路径, 如图2所示, CeO2的氧空位能捕捉气相中的氧形成超氧自由基(
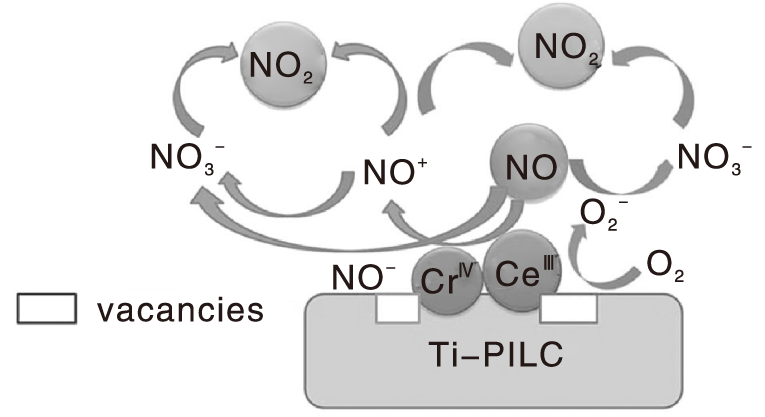 | 图2 Ce改性Cr/Ti-PILC催化剂上NO氧化反应路径Figure 2 Reaction pathways of NO oxidation over cerium modified Cr/Ti-PILC catalyst |
对于碳材料, Mochida I等[80]提出[NO-O-NO2]ad是形成NO2的关键中间体。Zhang Zhanquan等[92]认为大孔活性炭纤维上NO催化氧化的机理分两个步骤:(1)在大孔中气态NO和O2形成NO2, 而
总的来说, 不同催化剂参与的NO氧化的反应机理, 遵循E-R机理或者L-H机理或者两者的结合, 硝酸盐中间体在NO氧化反应中起到重要的作用。
在废气中, 不可避免有水的存在, 因而水对NO氧化的影响是很重要的研究内容之一。水对NO氧化有负面影响, 主要是因为水的存在会形成硝酸盐物种, 阻碍了催化剂表面对NO的吸附。硝酸盐物种的形成是由于反应生成的NO2和H2O形成硝酸[93], HNO3与催化剂活性组分反应产生硝酸盐, 沉积在催化剂表面。Li Landong等[8]发现水蒸气对Pt/TiO2催化剂活性有轻微的抑制作用, 水蒸气浓度为质量分数2%时, 在反应温度为225 ℃时可减少10%的NO转化率, 增加水蒸气的浓度至质量分数5%, 对NO氧化的影响略大。Li Kai等[61]也评价了H2O对Mn-Co-Ce-Ox催化剂活性的影响, 水蒸气浓度为质量分数15%时, NO转化率降低15%, 在反应温度为150 ℃时, 撤除H2O之后, 活性完全恢复, 表明了H2O对NO氧化的抑制作用是可逆的, 而且H2O的影响可能在温度升高到270 ℃时消失[94]。Long R等[95]发现Mn基过渡金属氧化物在水蒸气温度(140~180) ℃时, 具有较好的抗水性能, 原因可能是催化剂表面存在高活性和氧化还原电位的Mnn+。Chen Hu等[96]也证实了水蒸气浓度为质量分数10%时, γ -MnO2催化剂显示出非常好的抗水性能。
SO2的毒化作用是NO催化氧化很严重的问题之一, SO2氧化成SO3, 而SO3不仅会占据催化剂吸附NO的活性中心, 而且会在催化剂表面形成硫酸盐物种, 从而降低NO催化氧化的活性[94, 97]。除此之外, SO2的引入也会改变硝酸盐形成的种类[8, 88]。Lin Fawei等[98]制备了一系列CeO2-MnOx催化剂用于NO氧化, 在SO2存在的情况下, NO转化率在400 min内从92%降低到22%。Tang Nian等[90]也提到了在SO2存在下, MnOx/TiO2催化剂上的催化活性降低, 而且在撤除SO2后, 活性也不能恢复到初始水平, 因为SO2能以硫酸盐的形式永久占据Mn活性位。Chen Zhihang等[99]发现Cr掺杂MnOx催化剂的抗硫性和催化活性都能得到很大提高, 因为催化剂中Cr与Mn之间存在的较高的氧化还原电位。Zhang Lei等[100]报道了MnxC
在过去的几十年间, 研究合适的NO氧化催化剂得到广泛关注, 本文主要总结了4种类型的催化剂, 包括负载型, 多元金属氧化物, 钙钛矿型以及碳基催化剂。到目前为止, 贵金属催化剂, 尤其是Pt催化剂是典型的NO氧化催化剂, 表现出卓越的催化性能。金属氧化物催化剂得到越来越多的关注, 由于其低成本, 高催化活性和稳定性。通过掺杂其它金属氧化物来对贵金属或金属氧化物催化剂进行改性被证实可以提高催化活性, 可归因为协同作用。碳基催化剂可用于常温下NO的氧化。另外, 不同催化剂上NO氧化的反应机理主要遵循E-R机理或L-H机理, 或者这两者机理的结合。
尽管NO氧化已研究出较多的催化剂, 但是仍然需继续研究获得更高活性的催化剂, 并且通过掺杂其它合适的金属氧化物能提高催化活性的机理需更深入的研究。分子筛催化剂由于其较大的比表面积, 大量的酸中心, 以及卓越的吸附性能, 显示出较高的催化活性, 而应用于NO氧化报道的文献相对较少, 在未来应该对于这类催化剂进行更深入的研究。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|