{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
原位漫反射红外光谱研究Ni-Fe/γ-Al2O3催化CO甲烷化反应
[武瑞芳, 王宁, 周翔, 王永钊*  , 赵永祥]
, 赵永祥]
, 赵永祥]
|
|


作者简介:武瑞芳,1985年生,女,山西省临县人,硕士,实验师,研究方向为多相催化。
利用原位漫反射红外光谱研究Ni/γ-Al2O3、Fe/γ-Al2O3和Ni-Fe/γ-Al2O3催化CO甲烷化反应。结果表明,CO和CO-H2混合气在Ni/γ-Al2O3和Ni-Fe/γ-Al2O3催化剂上均存在化学吸附,且Ni-Fe/γ-Al2O3对CO和CO-H2混合气的吸附能力明显强于Ni/γ-Al2O3催化剂,但在Fe/γ-Al2O3上未发生化学吸附。CO-H2混合气吸附时,Ni/γ-Al2O3表面以镍羰基氢化物吸附为主,而Ni-Fe/γ-Al2O3表面则以镍多氢羰基氢化物为主,特别是桥式镍多氢羰基氢化物。与镍羰基氢化物相比,镍多氢羰基氢化物结构中含有更多的氢原子,其较强的给电子作用提高了Ni中心对CO分子反键π轨道反馈电子的能力,导致Ni-C键增强,C-O键削弱。因此,与Ni/γ-
The catalytic performances of Ni/γ-Al2O3,Fe/γ-Al2O3 and Ni-Fe/γ-Al2O3 for CO methanation were studied by in-situ diffuse reflectance infrared spectroscopy.Results show that CO and CO-H2 mixed gas exhibit chemical adsorption on both Ni/γ-Al2O3 and Ni-Fe/γ-Al2O3 catalysts,and the adsorption capacity of Ni-Fe/γ-Al2O3 for CO and CO-H2 mixed gas is significantly stronger than that of Ni/γ-Al2O3,yet no chemical adsorption occurs on Fe/γ-Al2O3.When CO-H2 mixed gas is adsorbed,nickel carbonyl hydride exists on the surface of Ni/γ-Al2O3,while nickel multihydrogen carbonyl hydride,especially bridged nickel multihydrogen carbonyl hydride exists on the surface of Ni-Fe/γ-Al2O3.Compared with nickel carbonyl hydride,nickel multihydrogen carbonyl hydride contains more hydrogen atoms,whose stronger electron-donating property improves the ability of the Ni atom for feeding back electrons to the anti-bonded π orbital of CO molecular,resulting in that the Ni-C bond becomes stronger,and the C-O bond becomes weaker.Therefore,the C-O bond of CO molecular on the Ni-Fe/γ-Al2O3 catalyst breaks more easily and is hydrogenated to methane comparing with the Ni/γ-Al2O3 catalyst,which may be the direct reason why the Ni-Fe/γ-Al2O3 catalyst shows higher catalytic activity.
近年来, 质子交换膜燃料电池因其环境友好、操作温度低且工作效率高而受到关注[1, 2, 3, 4]。氢气是质子交换膜燃料电池的燃料, 主要通过蒸汽重整和碳氢化合物的部分氧化等途径制得[5, 6, 7], 但上述反应制得的氢气中不可避免地含有体积分数1%~2%的CO, 极易毒化燃料电池的电极[8, 9, 10]。富氢气体中微量CO的脱除广泛采用CO甲烷化法, 在报道的众多CO甲烷化催化剂中, Ni基催化剂因其催化活性高, 选择性好以及成本相对低廉等优势受到研究者的青睐[11]。
在CO甲烷化反应中, 双金属Ni基催化剂显示出比单金属催化剂更高的催化活性和稳定性[12, 13, 14, 15, 16, 17]。本课题组前期研究工作表明, 与Ni/γ -Al2O3和Fe/γ -Al2O3催化剂相比, Ni-Fe/γ -Al2O3双金属催化剂表现出更优异的CO甲烷化催化性能。在相同反应条件下, 220 ℃时Fe/γ -Al2O3催化剂未表现出CO甲烷化催化活性, Ni/γ -Al2O3催化剂上CO转化率仅为48%, 而Ni-Fe/γ -Al2O3双金属催化剂上CO则完全甲烷化。通过XRD、N2物理吸附-脱附、H2-TPR、H2-TPD等表征结果显示, 3种催化剂的织构性质并无明显差异, 但Ni-Fe/γ -Al2O3催化剂还原后形成Ni-Fe合金, H2吸附量明显增加[18]。为了进一步探究Ni/γ -Al2O3、Fe/γ -Al2O3和Ni-Fe/γ -Al2O3催化剂催化活性差异的原因, 本文采用原位漫反射红外光谱对3种催化剂上CO、CO-H2混合气的吸附行为进行比较研究, 旨在阐明催化剂上吸附物种的演变规律, 为高性能CO甲烷化催化剂的研发提供理论参考。
称取一定量Ni(NO3)2· 6H2O和Fe(NO3)2· 9H2O配制成单一盐水溶液和一定计量比的混合盐水溶液, 然后分别等体积浸渍于γ -Al2O3载体上[(40~60)目, 比表面积253 m2· g-1], 充分搅拌并静置0.5 h后, 120 ℃干燥3 h, 空气气氛400 ℃焙烧3 h, H2气氛400 ℃还原3 h, H2流速30 mL· min-1。所得催化剂为负载质量分数10%Ni/γ -Al2O3、10%Fe/γ -Al2O3和6%Ni-4%Fe/γ -Al2O3, 分别标记为Ni/γ -Al2O3、Fe/γ -Al2O3和Ni-Fe/γ -Al2O3催化剂。
原位漫反射红外光谱实验采用HVC-DRP-3型高温漫反射池, 在德国布鲁克公司TENSOR27型傅里叶变换红外光谱仪上进行。将一定量的催化剂粉末装入样品池, 并刮平样品表面, 以利于增强漫反射信号。
样品池首先通入氦气(流速20 mL· min-1)并逐渐升温至350 ℃, 恒温30 min以吹尽催化剂表面的空气。然后降至室温, 切换为常压氢气(流速为30 mL· min-1)并程序升温(2 ℃· min-1)至400 ℃, 在此温度下还原催化剂1 h。还原结束后切换为氦气吹扫30 min, 降至室温后切换为CO或体积分数0.5%CO-H2混合气, 控制流速为15 mL· min-1, 使气体在催化剂表面进行动态吸附, 然后每隔一定时间进行红外扫描, 扫描次数为128次, 分辨率为4 cm-1。
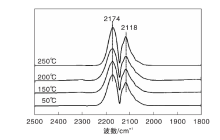
图1为不同温度下Fe/γ -Al2O3催化剂上CO吸附的原位漫反射红外光谱图。由图1可以看出, Fe/γ -Al2O3催化剂在2 174 cm-1和2 118 cm-1处出现气相CO的吸收峰[19], 除此之外, 未观察到其他红外吸收峰。
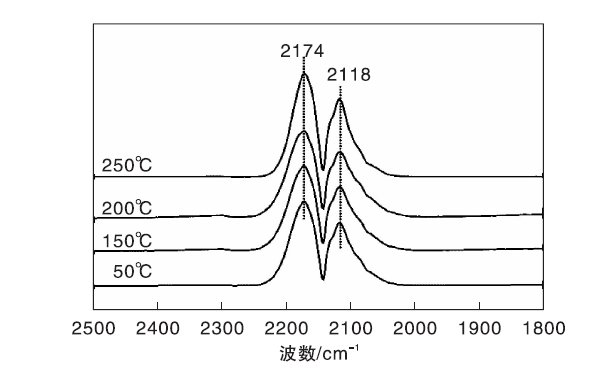 | 图1 不同温度下Fe/γ -Al2O3催化剂上CO吸附的原位漫反射红外光谱图Figure 1 In-situ DRIFTS of CO adsorbed on Fe/γ -Al2O3 catalyst at different temperatures |
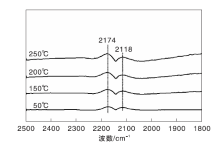
图2为不同温度下Fe/γ -Al2O3催化剂上CO-H2混合气吸附的原位漫反射红外光谱图。与图1相比, 气相CO的吸收峰强度明显减弱, 这是由于混合气中CO的体积分数较小所致。此外, 该催化剂上未观察到其他任何吸收峰。显然, 无论H2存在与否, Fe/γ -Al2O3催化剂均未表现出CO吸附行为, 这是其不具有CO甲烷化催化活性的直接原因。
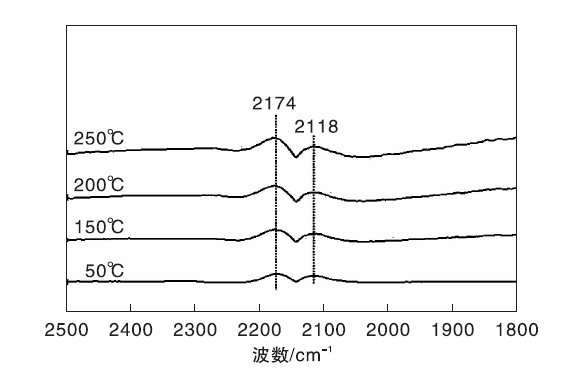 | 图2 不同温度下Fe/γ -Al2O3催化剂上CO-H2混合气吸附的原位漫反射红外光谱图Figure 2 In-situ DRIFTS of CO and CO-H2 mixed gas adsorbed on Fe/γ -Al2O3 catalyst at different temperatures |
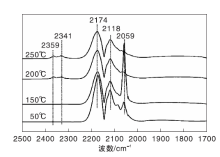
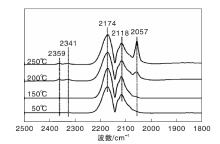
图3为不同温度下Ni/γ -Al2O3催化剂上CO吸附的原位漫反射红外光谱图。由图3可以看出, 50 ℃时Ni/γ -Al2O3催化剂表面除了气相CO吸收峰外, 在2 059 cm-1处还出现明显的线式CO(COL)吸收峰[19, 20, 21]。随着温度升高, COL峰强度明显增强, 但在110 ℃后开始呈下降趋势。200 ℃时, 在2 359 cm-1和2 341 cm-1处出现气相CO2的吸收峰, 表明随着温度升高, 部分化学吸附的CO发生歧化反应[22]。在(110~250) ℃, COL峰强度随着温度的升高逐渐减弱, 表明CO在Ni/γ -Al2O3催化剂表面化学吸附变弱。
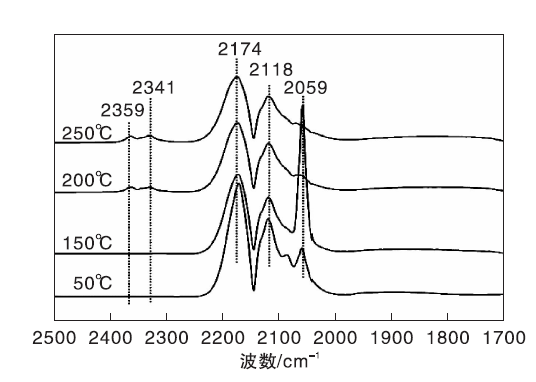 | 图3 不同温度下Ni/γ -Al2O3催化剂上CO吸附的原位漫反射红外光谱图Figure 3 In-situ DRIFTS of CO adsorbed on Ni/γ -Al2O3 catalyst at different temperatures |
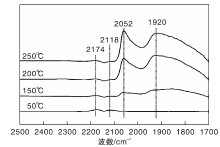
图4为不同温度下Ni/γ -Al2O3催化剂上CO-H2混合气吸附的原位漫反射红外光谱图。
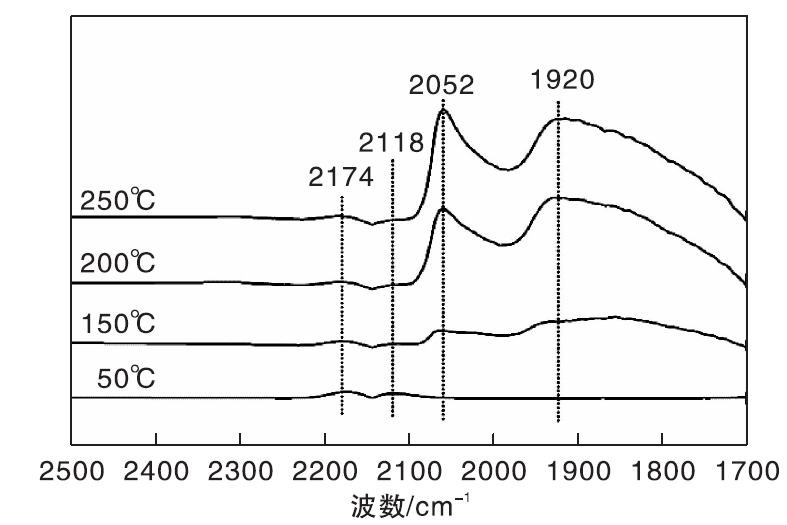 | 图4 不同温度下Ni/γ -Al2O3催化剂上CO-H2混合气吸附的原位漫反射红外光谱图Figure 4 In-situ DRIFTS of CO-H2 mixed gas adsorbed on Ni/γ -Al2O3 catalyst at different temperatures |
与图3相比较, 50 ℃下Ni/γ -Al2O3催化剂在2 059 cm-1处无明显的COL吸收峰, 其原因可能是催化剂表面活性位点优先被吸附的H2占据。150 ℃时, 2 052 cm-1和1 920 cm-1处出现两个新吸收峰, 与图4中的COL相比(2 059 cm-1), 2 052 cm-1处的吸收峰位置向低波数位移, 这是由于催化剂表面形成线式镍羰基氢化物( 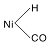
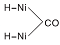
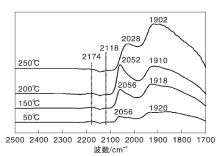
图5为不同温度下Ni-Fe/γ -Al2O3催化剂上CO吸附的原位漫反射红外光谱图。由图5可知, 50 ℃时除了气相CO吸附峰外, 在2 057 cm-1处也出现微弱的COL吸收峰, 且峰强度随着温度的升高逐渐增大, 这与CO在Ni/γ -Al2O3催化剂上的吸附行为明显不同, 后者的COL吸收峰强度随温度升高先增大后减小。可见, 与Ni/γ -Al2O3催化剂相比, Ni-Fe/γ -Al2O3催化剂表面存在较强的CO化学吸附中心, 与Ni-Fe/γ -Al2O3催化剂中Ni-Fe合金的形成有关[18]。
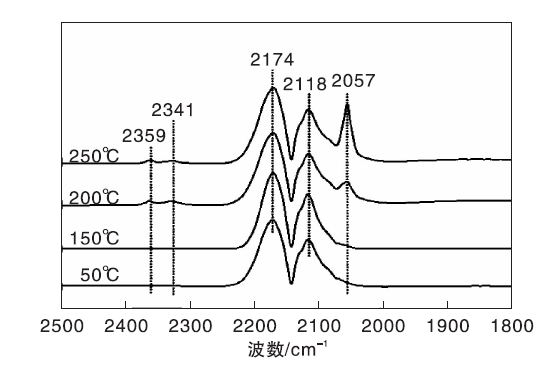 | 图5 不同温度下Ni-Fe/γ -Al2O3催化剂上CO吸附的原位漫反射红外光谱图Figure 5 In-situ DRIFTS of CO adsorbed on Ni-Fe/γ -Al2O3 catalyst at different temperatures |
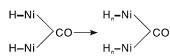
图6为不同温度下Ni-Fe/γ -Al2O3催化剂上CO-H2混合气吸附的原位漫反射红外光谱图。由图6可以看出, 50 ℃时Ni-Fe/γ -Al2O3催化剂在2 056 cm-1和1 920 cm-1处出现明显的线式和桥式镍羰基氢化物的吸收峰, 与图5进行对比, 同样证实H2的存在促进催化剂表面新吸附物种的产生。随着温度升高, Ni-Fe/γ -Al2O3催化剂表面线式和桥式镍羰基氢化物的吸收峰向低波数位移, 在250 ℃时分别位移至2 028 cm-1和1 902 cm-1处。这种位移现象可归因于线式和桥式镍羰基氢化物分别转化为相应的镍多氢羰基氢化物( 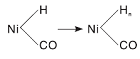
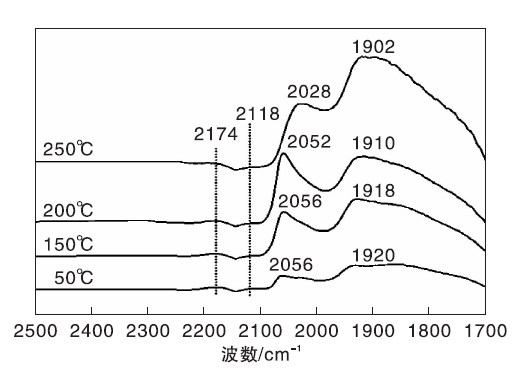 | 图6 不同温度下Ni-Fe/γ -Al2O3催化剂上CO-H2混合气吸附的原位漫反射红外光谱图Figure 6 In-situ DRIFTS of CO-H2 mixed gas adsorbed on Ni-Fe/γ -Al2O3 catalyst at different temperatures |
(1) 在CO吸附条件下, Fe/γ -Al2O3催化剂上未发生CO化学吸附, Ni/γ -Al2O3和Ni-Fe/γ -Al2O3催化剂上均有线式CO形成, 但后者对CO的吸附能力明显强于前者。
(2) 在CO-H2混合气吸附条件下, Ni/γ -Al2O3催化剂表面以镍羰基氢化物吸附为主, 而Ni-Fe/γ -Al2O3催化剂表面则以镍多氢羰基氢化物吸附为主, 特别是桥式镍多氢羰基氢化物。
(3) 镍多氢羰基氢化物的Ni中心对CO分子反键π 轨道更强的反馈电子能力导致Ni-C键增强, C-O键削弱, 更有利于C-O键断裂进而加氢进行甲烷化反应, 因而Ni-Fe/γ -Al2O3催化剂显示出更高的CO甲烷化催化活性。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|