

{kind=link}
{kind=link}
{kind=link}
{kind=link}
NaH催化合成3,4-二氢-2 H-吡嗪并[2,1-b]喹唑啉-1,6-二酮及其抗肝癌细胞活性评价
[葛维娟1 , 孟歌2, 3, *  , 谢紫轩
, 谢紫轩2 , 陈萍2 , 白宏1 ]
, 谢紫轩|
|


作者简介:葛维娟,1988年生,女,陕西省咸阳市永寿县人,硕士,讲师,研究方向为药物化学。
以邻氨基苯甲酰胺(5)为原料,依次经分子间环合、烷基化、氨解和分子内环合四步反应成功合成目标化合物3,4-二氢-2 H-吡嗪并[2,1-b]喹唑啉-1,6-二酮(10),总收率为46.5%,其中,中间体(8)、(9)及目标化合物(10)均未见文献报道,其结构经1H NMR和MS(ESI)确证,并采用MTT法初步评价目标化合物的体外抗肝癌活性。结果表明,3,4-二氢-2 H-吡嗪并[2,1-b]喹唑啉-1,6-二酮对SMMC-7721具有明显的抑制活性,优于阳性对照药舒尼替尼,可为进一步发现新型抗肝癌药物提供先导结构,也为该类衍生物的大量合成和结构改造提供参考方法。
The target compound 3,4-dihydro-2 H-pyrazino[2,1-b]quinazoline-1,6-dione(10) was successfully synthesized from anthranilamide(5) through four steps of intermolecular cyclization,alkylation,ammonolysis and intramolecular cyclization,the total yield of compound (10) was 46.5%.Intermediate (8),(9) and target compound (10) had not been reported,and their structures were confirmed by1H NMR and MS(ESI).The MTT assay was performed to evaluate anti-hepatoma activities of target compound.The results showed that compound (10) had significant inhibitory activity on SMMC-7721,which was better than the positive-drug sunitinib.Compound (10) can provide a lead structure for the further discovery of novel anti-hepatoma drugs,and also provide a reference for the massive synthesis and structural modification of these derivatives.
稠合氮杂环普遍存在于多种天然产物中, 在制药工业和材料科学中发挥着重要作用[1, 2]。特别是稠合的喹唑啉-4-酮是制备大量具有显著生物活性的生物碱类化合物的重要骨架[3, 4]。大约有150种天然的生物碱类化合物中含有稠合的喹唑啉-4-酮结构片段, 其中很多稠环结构具有抗肿瘤活性, 如色胺酮(Tryptanthrine)[5]、达尼喹酮(Batracyclin)[6]、鸭嘴花碱酮(Vasicinone)[7]、骆驼宁碱A(Luotonin A)[8]、吴茱萸次碱(Ruteacarpine)和吴茱萸碱(Evodiamine)[9]等。这些化合物具有类似的三环稠和喹唑啉杂环体系, 包括A、B和C环, 它们共有的主要结构骨架就是2, 3-稠合的(3H)-喹唑啉-4-酮, 结构如图1所示。
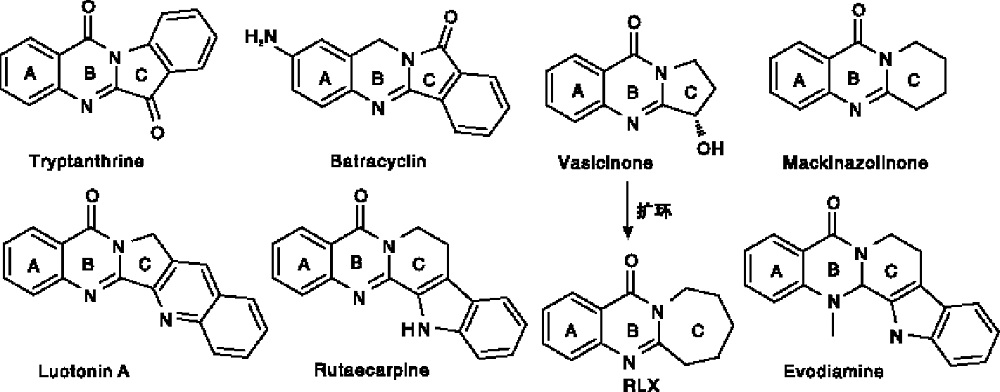 | 图1 具有稠合(3H)-喹唑啉-4-酮结构片段的代表性生物活性化合物Figure1 Representative bioactive compounds bearing fused quinazolin-4(3H)-ones as a structural fragment |
虽然2, 3-稠合(3H)-喹唑啉-4-酮类化合物是许多天然产物结构改造的核心, 但由于合成难度较大, 对此类环体系的合成及活性研究较少。例如, 法国科学家Thierry Besson课题组[10]曾试图以2-氨基苯甲酸(1)为原料, 通过特殊试剂Appel盐(4, 5-二氯-1, 2, 3-二硫代鎓盐氯化物, 2)合成所设计的目标化合物3, 4-二氢-2H-吡嗪并[2, 1-b]喹唑啉-1, 6-二酮(10), 却只得到了亚氨基取代产物(4), 没能如期获得目标化合物, 合成路线如图2所示。
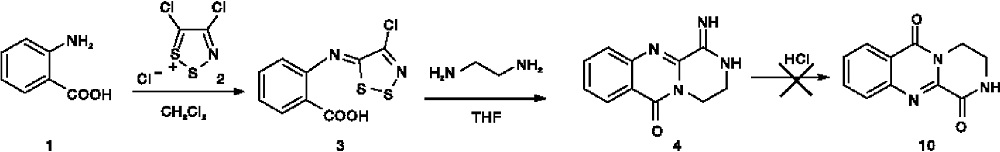 | 图2 Thierry Besson课题组假设的化合物(10)的合成路线Figure2 Hypothetic synthetic route of compound (10) by Thierry Besson's research group |
本文经过反复探究, 设计了一种合成化合物(10)的新方法, 合成路线如图3所示。以邻氨基苯甲酰胺(5)为原料, 通过与草酸二乙酯经分子间环合得到中间体(6)[11], 中间体(6)经二溴乙烷烷基化反应得到中间体(8)[12], 中间体(8)经乙酯基的氨解反应得到中间体酰胺(9)[13], 中间体酰胺(9)经分子内环合得到目标产物(10)[14]。所有新化合物结构经1H NMR和MS(ESI)确证, 并采用MTT法对目标物进行初步的抗肝癌细胞活性研究。
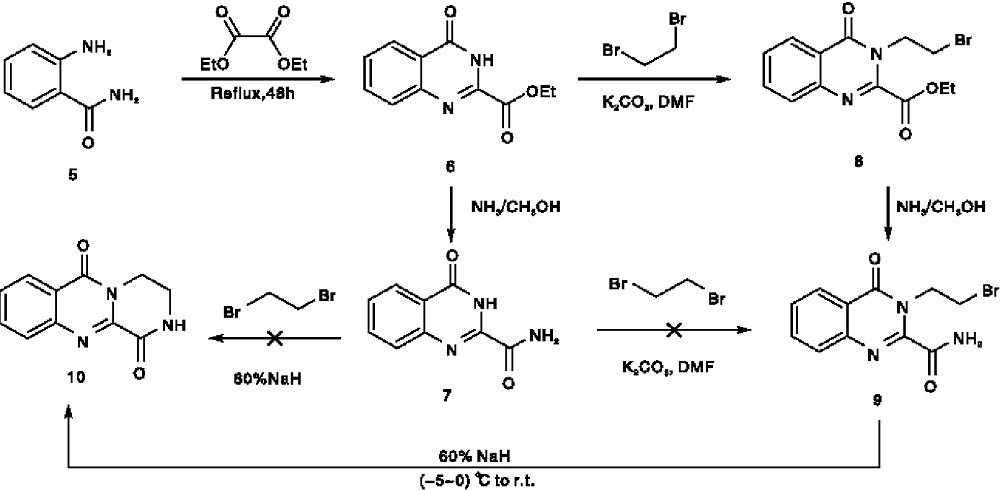 | 图3 化合物(10)的合成路线Figure 3 Synthetic route of compound (10) |
德国布鲁克公司AVANCF-300 MHz型核磁共振仪, DMSO-d6或CDCl3为溶剂, TMS为内标; GC/MS-QP2010气-质联用仪; XT-4型双目显微熔点仪; 酶标仪; 二氧化碳培养箱。
邻氨基苯甲酰胺、草酸二乙酯、二溴乙烷、氨甲醇溶液(7 N)、氢化钠、N, N-二甲基甲酰胺、石油醚、乙酸乙酯、碳酸钾、浓盐酸、四氢呋喃, 所用试剂和溶剂均为分析纯。
人肝癌细胞(SM-7721), 舒尼替尼阳性对照药。
1.2.1 (6)的合成
将草酸二乙酯50 mL(0.366 mol)分批加入融化的邻氨基苯甲酰胺(5)6.8 g(0.05 mol)中, 回流反应48 h(TLC跟踪)。反应完全后, 减压蒸干过量的草酸二乙酯。所得白色固体用冷的乙醇洗涤, 过滤后滤饼在冷乙醚中搅拌0.5 h, 过滤得细小白色针状晶体(6)8.6 g, 收率78.9%, 熔点(190~191) ℃ [文献[11]为(190~192) ℃]。
1.2.2 (8)的合成
称取(6)2.18 g(10.0 mmol)和碳酸钾1.52 g(11.0 mmol), 溶解于干燥N, N-二甲基甲酰胺(50 mL)中, 室温搅拌下, 滴加1, 2-二溴乙烷2.06 g(11 mmol), 30 min滴加完毕, 室温反应4 h(TLC跟踪)。过滤除去碳酸钾, 并向滤液中加水200 mL, 用乙酸乙酯(3× 100 mL)萃取, 合并有机相, 无水硫酸钠干燥, 旋蒸得到淡黄色油状物。经硅胶柱层析[洗脱剂:V(石油醚):V(乙酸乙酯)=5:1)]纯化得无色油状物, 加入少量的乙醇和适量的水, 超声条件下析出白色固体(8)2.50 g, 收率76.9%, 熔点(73~74) ℃。1H NMR (CDCl3, 400 MHz) δ (ppm):8.32(d, J=7.9Hz, 1H), 7.81(d, J=3.5Hz, 2H), 7.59(dd, J=7.7, 4.3Hz, 1H), 4.64~4.45(m, 4H), 3.86(t, J=6.5Hz, 2H), 1.48(t, J=7.1Hz, 3H)。MS m/z calculated for C13H13BrN2O3 [M+H]+325.16, found 326.1。
1.2.3 (9)的合成
称取中间体(8)1.30 g(4.0 mmol)于一个50 mL的圆底烧瓶中, 分批加入氨的甲醇溶液8.0 mL (7 N), 室温搅拌过夜(TLC跟踪)。待反应完全后, 将反应液倾入冰水(40 mL)中搅拌10 min。用浓盐酸调节pH值为5~6, 得到大量白色沉淀, 抽滤并用水洗涤滤饼, 真空干燥得白色固体(9)1.04 g, 收率为88.14%, 熔点(171~173) ℃。1H NMR(CDCl3, 400 MHz)δ (ppm):8.32(d, J=8.0Hz, 1H), 7.81(t, J=7.6Hz, 1H), 7.72(d, J=8.1Hz, 1H), 7.59(t, J=7.5Hz, 1H), 7.52(s, 1H), 5.84(s, 1H), 4.97(t, J=6.5Hz, 2H), 3.90(t, J=6.5Hz, 2H)。MS(EI) m/z calculated for C11H10BrN3O2 [M+H]+. 296.12, found 216.1(MS-Br)。
1.2.4 (10)的合成
称取化合物(9)0.89 g(3.0 mmol)溶解于四氢呋喃(25 mL)中, 在(-5~0) ℃缓慢加入0.24 g(6.0 mmol)的60%NaH, 室温条件下搅拌1 h(TLC跟踪)。待反应完全后, 将混合物减压浓缩, 加入冰水淬灭反应, 过滤得到白色粗产物, 用冷水、石油醚依次洗涤滤饼得到目标化合物(10)0.56 g, 收率为86.8%, 熔点> 300 ℃。1H NMR(DMSO-d6, 400 MHz)δ (ppm):8.91(s, 1H), 8.18(d, J=7.7Hz, 1H), 7.89(t, J=7.2Hz, 1H), 7.82(d, J=8.0Hz, 1H), 7.63(t, J=7.4Hz, 1H), 4.22(m, 2H), 3.55(t, 2H)。MS m/z calculated for C11H9N3O2 [M+H]+ 215.21, found 215.1。
以舒尼替尼(sunitinib)为阳性对照药, 肝癌细胞系SMMC-7721作为受试细胞株, 采用MTT比色法初步测试目标化合物(10)体外抗肝癌细胞活性[15]。
肝癌细胞系SMMC-7721以含10%胎牛血清的DMEM培养基(Corning cat no. 10017236R), 37 ℃、5%CO2培养箱培养至对数生长期, 然后以0.02%EDTA-0.25%胰酶37 ℃消化2 min, 1 000 rpm离心5 min收集细胞, 制备1× 105/mL细胞悬液, 按100 μ L/孔将细胞接种于96孔细胞培养板, 37 ℃、5%CO2培养箱过夜培养细胞。将所测试的化合物(10)以二甲基亚砜配置为100 mmol· L-1贮存液, 分别按(0~50) mmol· L-1系列浓度梯度进行加药处理, 并设对照组, 均设3个复孔, 总体积200 μ L/孔, 继续37 ℃、5%CO2培养细胞24 h、48 h、72 h。每孔加入20 μ L/孔MTT(5 mg· mL-1), 继续培养(3~4) h, 移除培养液, 加150 μ L/孔二甲基亚砜, 室温下振荡15 min, 用酶标仪(λ =490 nm)测定待测液的吸光度值, 分别在24 h、48 h、72 h测定并记录产物对SMMC-7721细胞抑制活性。将抑制率与对应的药物浓度数据输入GraphPad Prism7.00软件中, 拟合抑制率变化曲线并计算半数抑制浓度(IC50)值。
目标化合物(10)三个环的构建经过四步完成。其中第一步按照文献报道的方法比较容易获得了中间体(6), 从而构建了A环和B环。该合成路线的新颖之处在于通过后续三个步骤构建C环。尽管每一步类似反应的具体方法和条件已在文献中报道, 但用于构建这种特殊骨架的整体合成路线尚未报道。这种新设计的合成路线是我们研究小组首次提出并成功验证, 并且已在其他相关稠环系统的合成中得到实践。
在探索这种合成路线的过程中, 也遇到许多困难。首先, 为了获得关键的中间体羧酰胺(9), 我们最初设计了另一种替代方法(见图3), 即将中间体(6)先经过氨甲醇氨解得到羧酰胺中间体(7), 然后尝试通过将(7)与1, 2-二溴乙烷进行常规烷基化来获得羧酰胺(9), 但无论用何种碱性催化剂(包括碳酸钾和氢化钠), 都无法得到所需烷基化产物。猜想羧酰胺(7)的惰性性质可能与喹唑啉上3-NH的正电子密度比羧酸酯(6)更高, 因为随后(6)通过烷基化试剂成功转化为中间体(8), 然后(8)经氨解又成功得到了中间体(9)。
合成路线的最后一步对我们研究小组来说是一项艰巨的任务, 就像Thierry Besson小组的相关类似研究工作一样。为了获得化合物(10), 最初尝试在室温下用碳酸钾对羧酰胺(9)进行分子内环化, 在连续反应15 h的TLC分析中未检测到任何新产物, 在油浴上升高反应温度也不能改善反应过程。经过反复试验, 考虑到催化碱的碱性可能太弱, 于是选择了碱性更强的氢化钠作为催化剂, 室温条件下反应1 h, 成功获得目标化合物(10), 总收率46.5%。受此结果鼓舞, 设想在相同的NaH条件下, 是否可以通过(7)和二溴乙烷之间的一步分子间缩合直接获得化合物(10), 但实验结果表明, 这种方法无法获得目标产物(图3)。
将化合物(10)通过配制成0 mmol· L-1、0.781 3 mmol· L-1、1.562 5 mmol· L-1、3.125 mmol· L-1、6.25 mmol· L-1、12.5 mmol· L-1、25 mmol· L-1、50 mmol· L-1共8个浓度, 分别处理细胞株24 h、48 h、72 h, 以舒尼替尼(sunitinib)为阳性对照药, 同法处理。采用MTT比色法测定其抗增殖作用, 结果如图4所示。 由图4可以看出, 肝癌细胞经8个不同浓度的化合物(10)作用24 h 后, 细胞生长均受到抑制, 但只有在浓度为50 mmol· L-1时, 其对肝癌细胞抑制率优于阳性对照组, 有极显著差异(P< 0.001)。化合物(10)与肝癌细胞作用48 h后, 浓度分别为25 mmol· L-1、50 mmol· L-1时, 其对肝癌细胞抑制率高于阳性对照组, 有极显著差异(P< 0.001)。化合物(10)与肝癌细胞作用72 h后, 在浓度为0.781 3 mmol· L-1时, 其对肝癌细胞抑制率高于阳性对照组, 有显著差异(P< 0.01); 在浓度为1.562 5 mmol· L-1时, 其对肝癌细胞抑制率高于阳性对照组, 存在极显著差异(P< 0.001); 在浓度为3.125 0 mmol· L-1时, 其对肝癌细胞抑制率高于阳性对照组, 有显著差异(P< 0.05); 而在其余的浓度条件下, 与对阳性对照组相比, 发现抑制率没有明显变化。
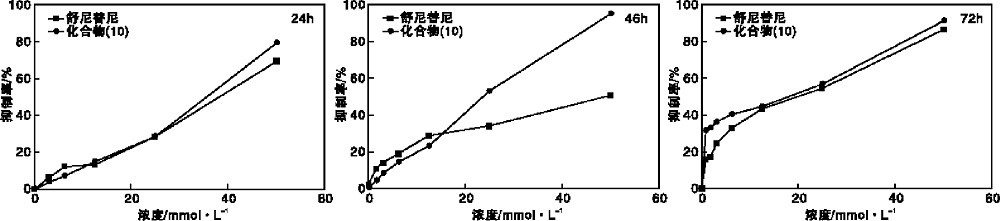 | 图4 化合物(10)与舒尼替尼对SMMC-7721细胞系的生物抑制活性对比图Figure 4 Biological inhibitory activity comparision of compound 10 with sunitinib against SMMC-7721 cell line |
在GraphPad Prism7.00软件中, 拟合抑制率变化曲线并计算了化合物(10)、舒尼替尼对SMMC-7721细胞系分别在24 h、48 h和72 h的半数抑制浓度(IC50)值, 结果如表1所示。由表1可以看出, 化合物(10)在24 h、48 h和72 h的IC50值均小于阳性对照组, 初步表明化合物(10)是一种很有前途的新型抗肝癌药物先导化合物。
| 表1 化合物(10)、舒尼替尼对SMMC-7721细胞系分别在24 h、48 h和72 h的IC50值(mmol· L-1) Table 1 The IC50 values of the compound (10), sunitinib on SMMC-7721 cell line at 24 h, 48 h and 72 h respectively(mmol· L-1) |
(1)以天然产物所共有的三环稠合喹唑啉结构模板, 开发了一种有效的构建2, 3-稠合的(3H)-喹唑啉-4-酮骨架的方法, 经过分子间环合、烷基化、氨解、分子内环合四步化学反应合成出一个结构新颖的3, 4-二氢-2H-吡嗪并[2, 1-b]喹唑啉-1, 6-二酮化合物(10), 并采用MTT法初步评价其体外抗肝癌活性。
(2)化合物(10)对肝癌细胞系SMMC-7721具有明显的抑制活性, 优于阳性对照药舒尼替尼, 为进一步发现新型抗肿瘤药物提供先导结构, 也为该类衍生物的大量合成和结构改造提供参考方法。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|