{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
咔唑类生物碱玫瑰树碱的合成研究进展
[韩宁娟1 , 黄新炜2 , 刘建利3, *  ]
]
]
|
|


作者简介:韩宁娟,1985年生,女,硕士,讲师,主要从事药物分析工作。
玫瑰树碱是从常绿植物椭圆玫瑰树中分离得到的具有平面结构的咔唑类生物碱,对DNA中的碱基对有特异性作用,该化合物及其水溶性衍生物具有抗肿瘤和抗HIV活性,且结构相对简单,因此,研究者开发了多种合成策略。本文按照构建环系的不同方式,综述玫瑰树碱化学合成方法,其中苯环吡咯环+吡啶环(AB+D环)构建苯环(C环)的合成策略,合成步骤相对较少,吲哚和吡啶衍生物原料较易得到,该策略具有更广阔的发展空间。
Ellipticine,an alkaloid isolated from the evergreen tree Ochrosia elliptica,is one of the simplest naturally carbazole alkaloids and has a planar structure.The preclinical experiments and clinical trials showed that this compound and several of its more soluble derivatives exhibited significant antitumor and anti-HIV activities.The excellent activities stimulated a strong interest in the synthesis of ellipticine and its analogues.A lot of synthetic strategies were developed to access this structural motif.In this paper,according to the different ways of constructing the ring system,the synthetic methods of ellipticine chemical synthesis are summarized.The AB+D ring is used to construct the C ring.The synthesis steps are relatively few and has broaden development space.
咔唑生物碱在药物开发中发挥着重要作用, 咔唑骨架本身被认为是鉴定新药效团的特殊结构[1]。玫瑰树碱是吡啶并[4, 3-b]咔唑生物碱最突出的代表[2], 1959年从澳洲野生常绿植物椭圆玫瑰树中分离得到[3]。临床前和临床试验表明该化合物及其水溶性衍生物显示显著的抗肿瘤和抗艾滋病活性, 且副作用有限。9-羟基玫瑰树碱、N-2-甲基-9-羟基玫瑰树碱醋酸盐已经在临床上用于粒细胞白血病、晚期乳腺癌和其他实体瘤的治疗[4]。抗癌作用机理是插入DNA双螺旋链中, 因为其结构与嘌呤-嘧啶碱基对类似, 与DNA拓扑异构酶II相互作用, 还与DNA共价结合形成新的第三种作用模式[5]。玫瑰树碱的季铵盐是D-丙氨酸-D-丙氨酸连接酶抑制剂。该酶是细菌细胞壁合成必须的胞内酶, 是极具吸引力的新型抗菌药的靶标[6]。因此, 玫瑰树碱在新型抗菌药的研发方面很有前景。
玫瑰树碱生物活性优异, 结构相对简单, 在生源关系、化学合成、药理作用、代谢过程等, 取得了很多有意义的结果。Sainsbury M等[7]综述了基于吡啶并咔唑骨架最终环形成的15条合成路线。Barone R等[8]报道了基于计算机产生的构建玫瑰树碱环系的合成策略。计算程序基于两个键切断的逆合成分析表明有253条合成路线。Hewlins M J E等[9]综述了包括玫瑰树碱的吡啶并咔唑及其类似物的合成。Gribble G W等[10]综述了玫瑰树碱及其相关的
吡啶并咔唑生物碱, 其中合成策略按照关键键的形成分类。Thompson D等[11]综述了玫瑰树碱的合成及生物活性。Kansal V K等[12]的综述主要集中在玫瑰树碱的生源、抗肿瘤活性及机理, 及一些重要的合成方法。Gribble G W[13]对包括玫瑰树碱的吡啶并咔唑生物碱的合成及生物活性进行综述。Langlois Y等[14]综述了玫瑰树碱及其同类化合物的抗肿瘤活性。Ä lvarez M等[15]在有关吲哚生物碱的专著中也提及了玫瑰树碱。Pindur U等[16]将作为脱氧核糖核酸嵌入剂的抗肿瘤活性药物嵌入复合物的分子模型, 在细胞抑制因子的分子水平上提出了与DNA和碱基对寡核苷酸嵌入法制备吡啶并咔唑生物碱。KnÖ lker H J等[17]在咔唑生物碱的分离及合成综述中也包括了玫瑰树碱及其类似物。
本文总结玫瑰树碱生源关系, 按照合成时构建环系的不同方式, 综述玫瑰树碱化学合成研究进展。
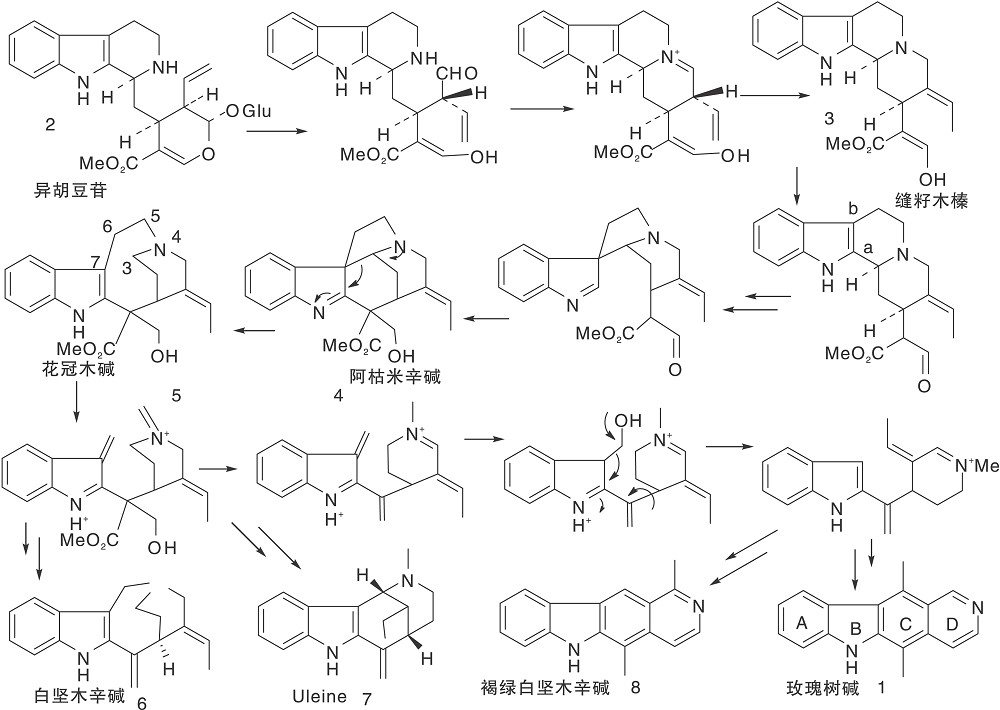
玫瑰树碱生源关系复杂[18], 如图1所示。
 | 图1 玫瑰树碱生源关系图Figure 1 Relationship between the alkaline sources of rose trees |
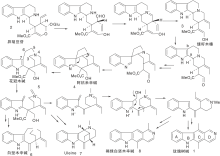
合成前期与其他单萜吲哚生物碱一样由色胺与裂环马钱素缩合生成异胡豆苷strictosidine(2); 经过糖苷水解、环化、还原等一系列过程转变成为缝籽木榛geissoschizine(3); 3-位碳由a 向b位移位、关环生成前阿枯米辛碱preakuammicine(4), 开环成花冠木碱stemmadenine(5); 后者经5, 6-位键断开、脱羧后, 一个方向是生成白坚木辛碱apparicine(6)和单萜吲哚生物碱[uleine(7), dasycarpidone, conolidine, ervaticine], 这些化合物的共同特点是吲哚环与4-氮之间只有一个碳连接。另一个方向是经氧化、脱甲醛、关环生成玫瑰树碱(1)和褐绿白坚木辛碱olivacine(8), 2者的共同特点是吲哚环与4-氮之间的两个碳均失去了, 因此, 这两个方向的化合物常被归为一大类。玫瑰树碱中A和C为苯环, B为吡咯环, D为吡啶环。
按照构建环系的不同方式, 玫瑰树碱合成可分为A+B+C+D, AB+C+D, AB+D, A+CD, ABC+D几种方式。
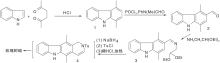
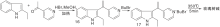
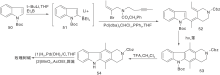
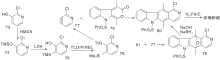
Woodward R B等[19]采用的最早的合成路线, 证实了玫瑰树碱结构。之后经过改进, 合成效率有很大提高[20], 至今仍被广泛应用。该合成路线采用吲哚与2, 5-己二酮在氯化氢催化下缩合形成C环, 再经甲酰化、胺化、还原、关环等步骤形成D环后得玫瑰树碱, 如图2所示。
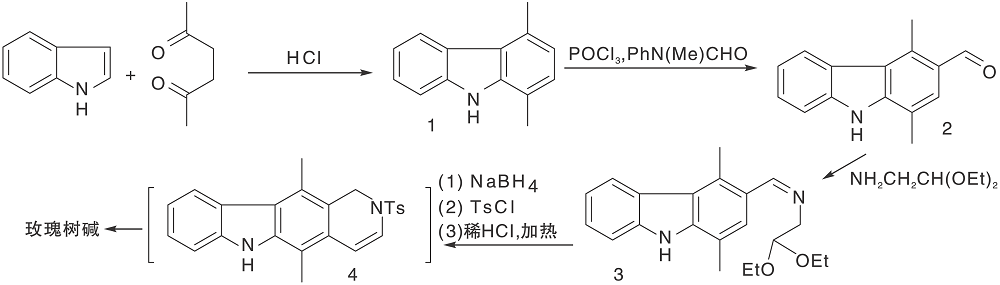 | 图2 玫瑰树碱经典合成路线Figure 2 Classic synthetic route of rosmarinine |
2.2.1 吲哚与吡啶衍生物形成C环
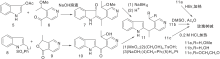
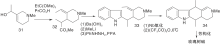
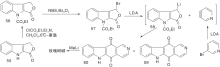
Sainsbury M等[21]报道了多条a-取代或b-取代的吲哚(5, 8)与取代吡啶缩合、还原、脱水、环化、空气氧化生成玫瑰树碱合成路线, 如图3所示, 吲哚与吡啶衍生物形成C环(Ⅰ )。氯化氢催化下, 吲哚与3-乙酰吡啶在乙醇溶液中加热、氢化还原生成12, 经锌粉乙酸酐环化得玫瑰树碱。也有报道吲哚格氏试剂与3-(1-氯乙基)吡啶经β -烷基化反应生成12。吡啶盐13上引入腈基后与甲基锂反应、酸化生成玫瑰树碱。中间体14也可由15与丙烯腈反应制得, 如图4所示, 吲哚与吡啶衍生物连接形成C环(Ⅱ )。
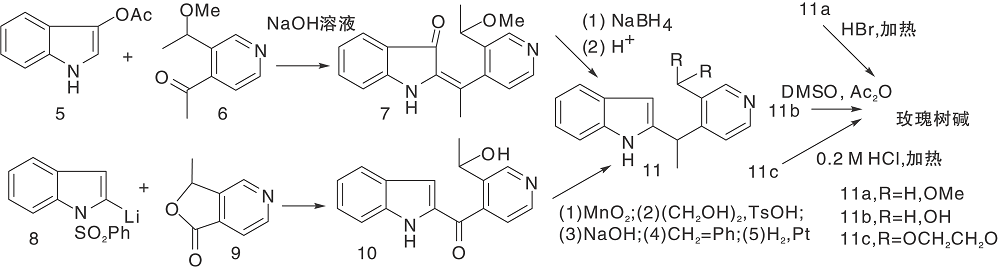 | 图3 吲哚与吡啶衍生物连接形成C环(Ⅰ )Figure 3 C-ring formed by the connection ofindole and pyridine derivatives (Ⅰ ) |
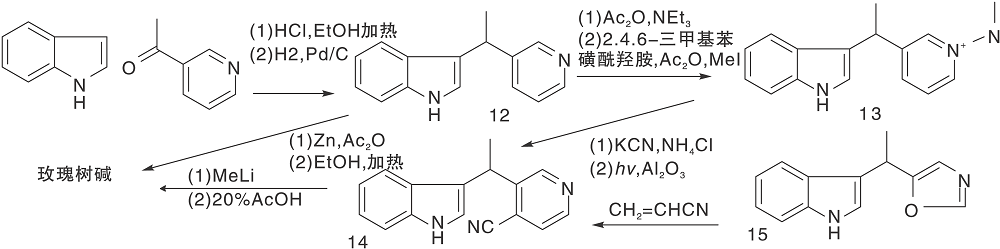 | 图4 吲哚与吡啶衍生物连接形成C环(Ⅱ )Figure 4 C-ring formed by the connection ofindole and pyridine derivatives (Ⅱ ) |
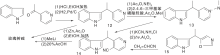
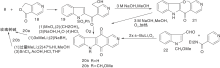
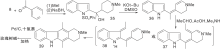
Bergman J等[22]报道了以热解为关键步骤构建C环的简便方法, 2-乙基吲哚与3-乙酰基吡啶缩合生成16, 再与溴丁烷反应生成吡啶盐17、热解环化、脱去丁烷生成玫瑰树碱, 如图5所示, 吲哚与吡啶衍生物连接形成C环(Ⅲ )。以吲哚8、22分别与吡啶衍生物缩合制成醌20a、20b, 然后与甲基锂反应、还原得玫瑰树碱[23], 如图6所示, 吲哚与吡啶衍生物连接形成C环(Ⅳ )。
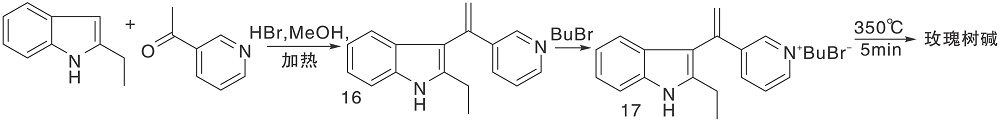 | 图5 吲哚与吡啶衍生物连接形成C环(Ⅲ )Figure 5 C-ring formed by the connection of indole and pyridine derivatives (Ⅲ ) |
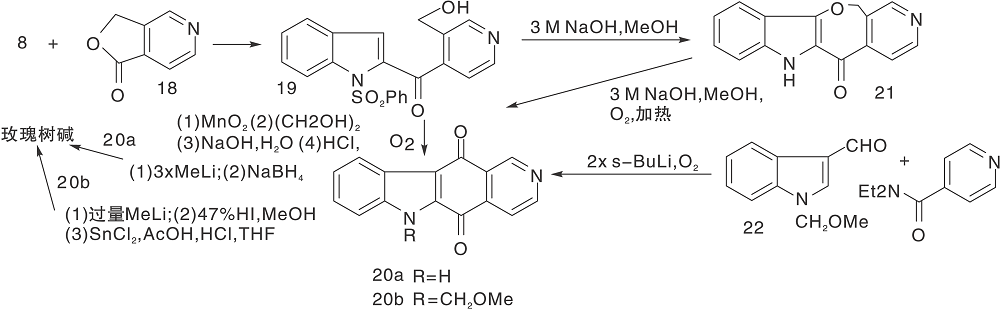 | 图6 吲哚与吡啶衍生物连接形成C环(Ⅳ )Figure 6 C-ring formed by the connection of indole and pyridine derivatives(Ⅳ ) |
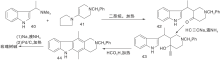
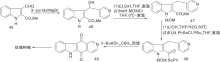
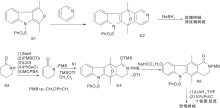
Zee S H等[24]报道了以热解为关键步骤的合成路线, 以吲哚与3-乙酰吡啶缩合形成23, 然后热解得24, 还原后在锌粉、醋酸酐作用下关环得27。脱乙酰基、氧化芳香化制备得到玫瑰树碱, 如图7所示, 吲哚与吡啶衍生物连接形成C环(Ⅴ )。Hibno S等[25]报道了另一条以热解为关键步骤的合成路线, 以二异丙基氨基锂(LDA)连接吲哚和吡啶衍生物28和29得到30, 热解得玫瑰树碱和褐绿白坚木辛碱, 如图8所示, 吲哚与吡啶衍生物连接形成C环(Ⅵ )。
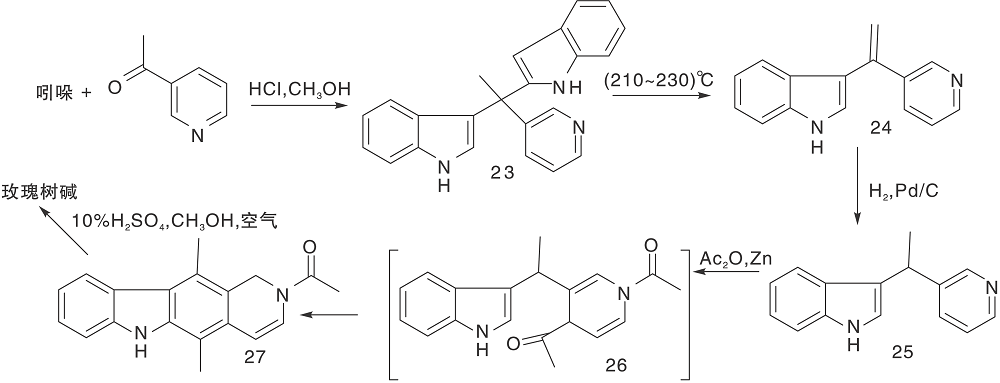 | 图7 吲哚与吡啶衍生物连接形成C环(Ⅴ )Figure 7 C-ring formed by the connection of indole and pyridine derivatives(Ⅴ ) |
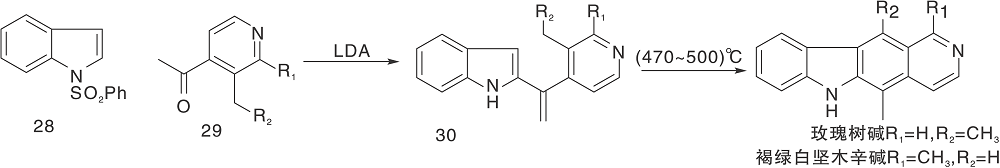 | 图8 吲哚与吡啶衍生物连接形成C环(Ⅵ )Figure 8 C-ring formed by the connection of indole and pyridine derivatives(Ⅵ ) |
2.2.2 采用部分还原的D环中间体构建C环
采用部分还原的D环构建玫瑰树碱骨架中的C环, 芳香化步骤在最后, 这是由于玫瑰树碱是稳定的芳香分子, 结构中无敏感基团, 可以剧烈脱氢。Langlois Y等[14]模拟生物合成步骤, 图9为模拟生物合成构建C环(Ⅰ ), 图10为模拟生物合成构建C环(Ⅱ )。
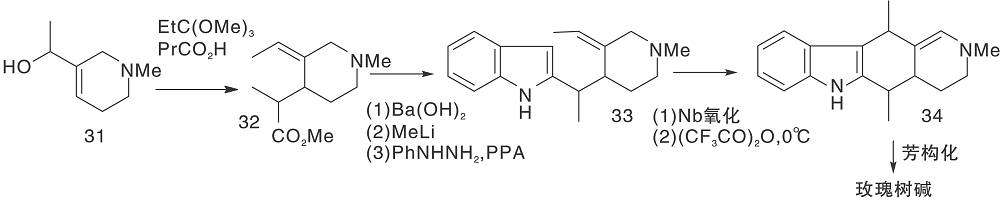 | 图9 模拟生物合成构建C环(Ⅰ )Figure 9 Construction of C-ring by simulated biosynthesis (Ⅰ ) |
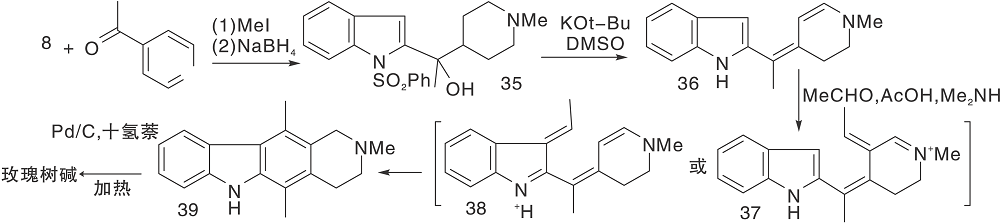 | 图10 模拟生物合成构建C环(Ⅱ )Figure 10 Construction of C-ring by simulated biosynthesis (Ⅱ ) |
图11所示合成路线先将吲哚(40)与部分还原的吡啶(41)偶联, 采用炔基加成引入D环的两个碳(43), 在一定的氧化条件下, 酸催化关环形成C环, 最后芳香化得到玫瑰树碱[26]。
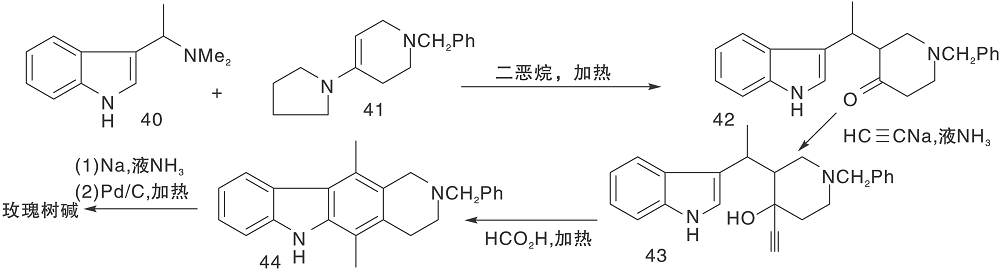 | 图11 吲哚与部分还原的吡啶构建C环Figure 11 Construction of C-ring by simulated biosynthesis |
2.2.3 自由基环化构建C环
自由基环化是构建杂环的方法, Bennasar M L等[27]利用吡啶格斯试剂与吲哚45反应, 然后还原、保护吲哚氮、水解、硒酯化、环化得到醌49。49与甲基锂反应得到玫瑰树碱, 如图12所示。应用类似的方法研究者还合成了玫瑰树碱的衍生物糖胺丁酸guatambuine和8。
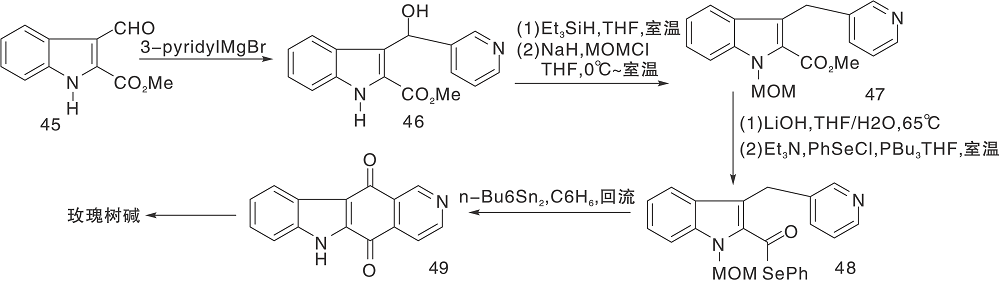 | 图12 自由基环化构建C环Figure 12 Construction of C-ring by free radical cyclization |
2.2.4 光环化构建CD环
Ishikura M等[28]报道了以吲哚硼酸酯(51)和溴丙烯衍生物为原料在钯催化下的串联偶合反应, 并经光环化、氧化芳香化反应制备得到玫瑰树碱, 如图13所示。同时也得到了一些结构类似的衍生物。
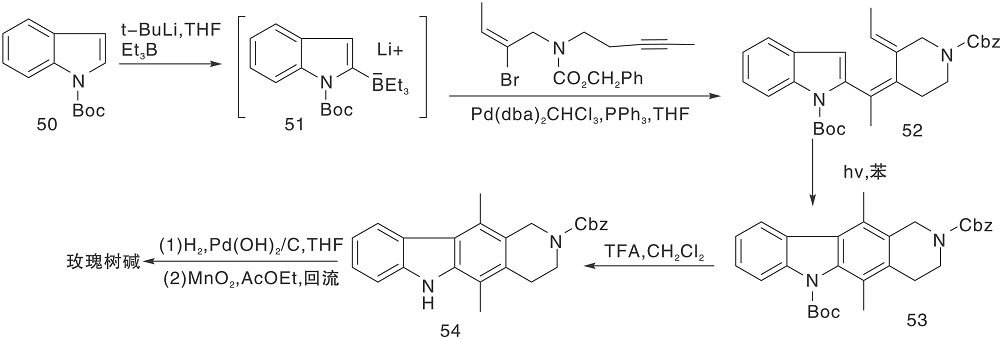 | 图13 光环化构建CD环Figure 13 Halo construction of CD ring |
2.2.5 D-A环加成反应构建C环
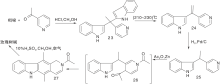
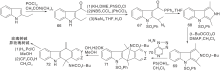
Mal D等[29]采用氮气保护吲哚内酯(55), 溴代后制成锂试剂(58)与3-溴吡啶制成的锂试剂经[4+2]环加成反应生成59和60, 再与甲基锂反应、还原得玫瑰树碱, 如图14所示, D-A环加成反应构建C环(Ⅰ )。利用该环加成反应可以合成多种衍生物。
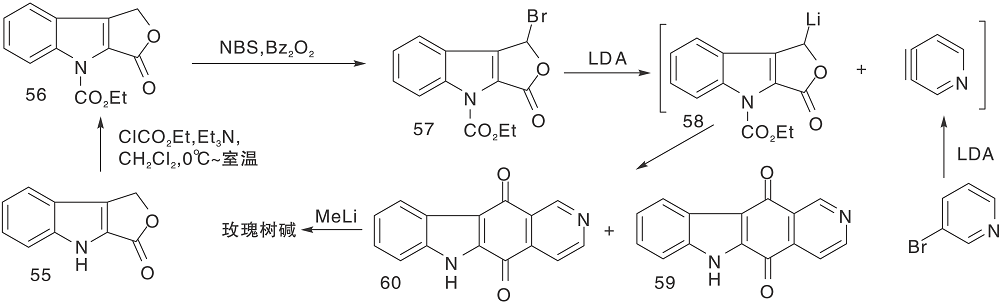 | 图14 D-A环加成反应构建C环(Ⅰ )Figure 14 Construction of C-ring by D-A cycloaddition reaction(Ⅰ ) |
Gribble G W等[30]采用吲哚并呋喃(61)与吡啶炔进行环加成反应制备得到62, 然后还原为玫瑰树碱和异玫瑰树碱; 采用对甲氧苯磺酰基代替61中的苯磺酰基也可以进行相应的反应[31], 为了选择性的得到玫瑰树碱, 采用二氢吡啶酮代替吡啶炔进行环加成反应得到64, 再经过转化制备得到目标化合物[32], 如图15所示, D-A环加成反应构建C环(Ⅱ )。
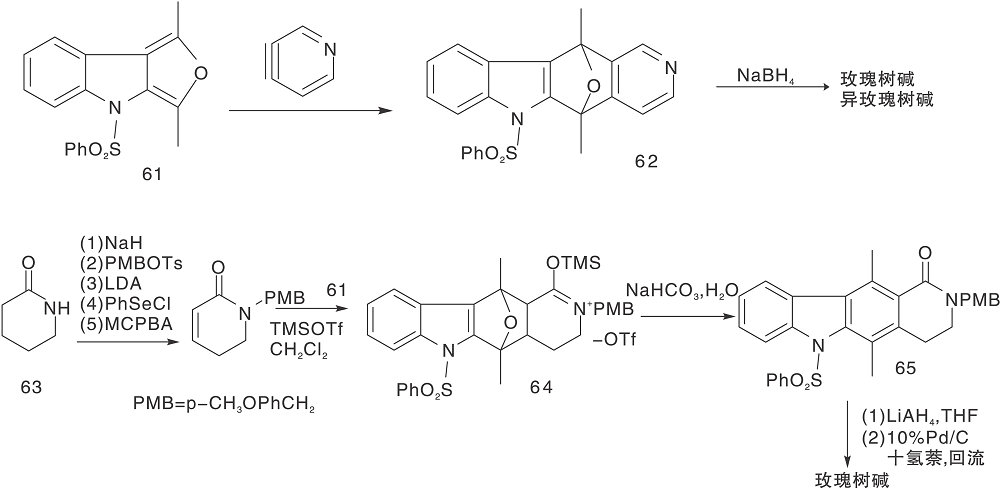 | 图15 D-A环加成反应构建C环(Ⅱ )Figure 15 Construction of C-ring by D-A cycloaddition reaction(Ⅱ ) |
Sha C K等[33]采用2-乙基吲哚, 经过酰化、取代、环化等反应步骤制备得到吲哚并吡咯(68), 再与吡啶炔的前体70进行D-A环加成反应生成71, 经过去保护、还原等反应步骤后生成玫瑰树碱及其异构体, 如图16所示, D-A环加成反应构建C环(Ⅲ )。
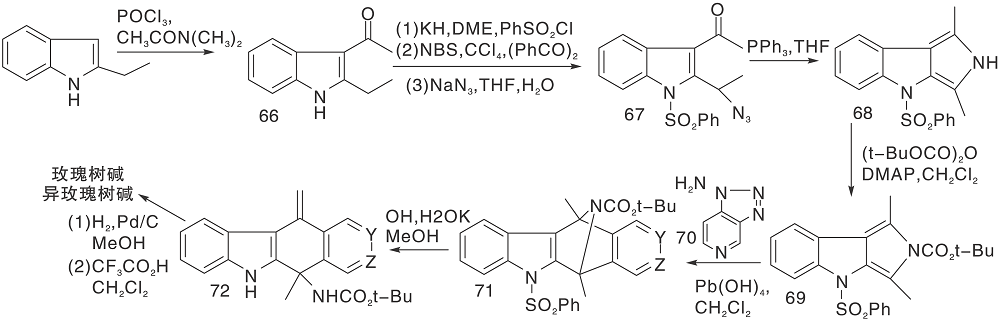 | 图16 D-A环加成反应构建C环(Ⅲ )Figure 16 Construction of C-ring by D-A cycloaddition reaction(Ⅲ ) |
Dí az M T等[34]采用用不同取代的吡啶炔与吲哚并呋喃(61)、吲哚并吡咯(68)及吲哚内酯(79)进行环加成反应合成玫瑰树碱及异玫瑰树碱, 如图17所示, D-A环加成反应构建C环(Ⅳ )。研究发现, 以61与氯代吡啶炔(77)反应合成玫瑰树碱, 区域选择性和产率均提高约6倍。
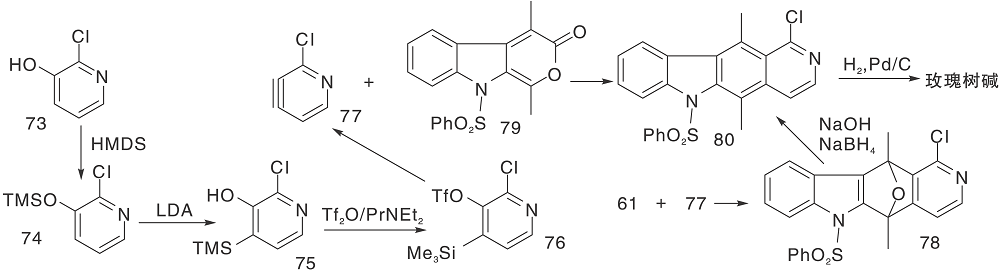 | 图17 D-A环加成反应构建C环(Ⅳ )Figure 17 Construction of C-ring by D-A cycloaddition reaction(Ⅳ ) |
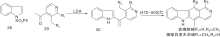
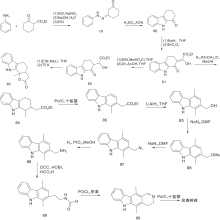
Dilek Ö 等[35]以苯胺、2-氧代环己烷羧酸乙酯为原料, 通过缩合、吲哚化、缩合、还原反应形成四氢咔唑衍生物, 后期D环环化制备得到玫瑰树碱, 如图18所示。合成路线的主要特征是在三氟乙酸介质中, 与四氢咔唑衍生物稠合反应形成内酯, 在对甲苯磺酸和钯碳催化下开环和芳构化反应制得玫瑰树碱。
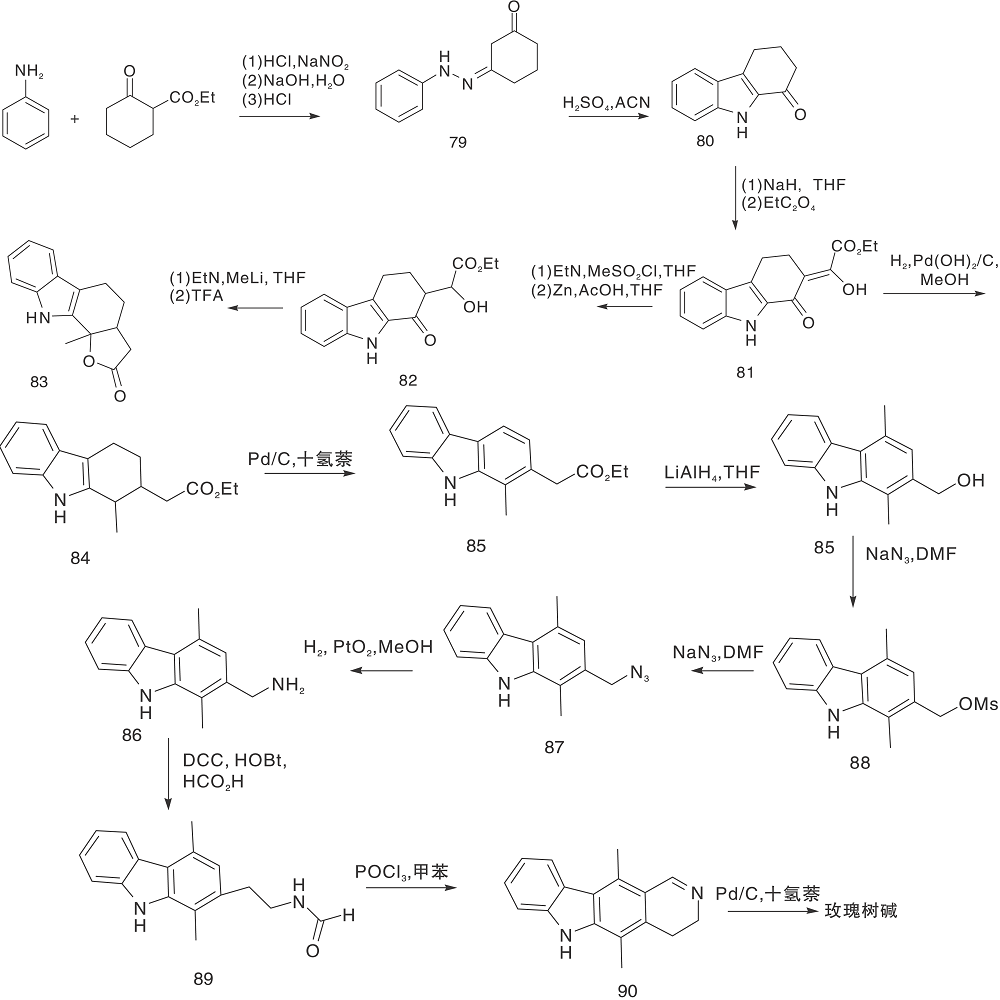 | 图18 玫瑰树碱的合成路线Figure 18 Synthetic route of rosacine |
(1) 以AB+D环构建C环的合成策略属于收敛性合成, 合成步骤相对较少。吲哚和吡啶衍生物较易得到, 因此该策略得到了广泛应用。
(2) 以A+CD构建B环的策略具有同样的性质, 但此路线中异喹啉类衍生物相对于吲哚来说不太常见, 往往需要自制, 使得此路线相对不易实现。
(3) A+D环同时构建BC环也是一个不错的思路, 但该反应所需的中间体结构、试剂在目前条件下不易得到, 还有待改进。
(4) 经典的AB+C+D合成策略至今还有应用是因为1本身并不很复杂, 线性合成策略所需合成步骤可以接受。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|