{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CO2与胺/H2(氢硅烷)催化转化为N-甲酰胺和N-甲基胺催化剂研究
[王静, 刘建芳, 冉真真, 季生福*  ]
]
]
|
|


作者简介:王 静,1995年生,女,在读硕士研究生,研究方向为纳米催化剂及反应。
随着工业化发展,大量的温室气体CO2被排入大气,对大气环境的影响越来越严重。但CO2也是一种廉价的碳源,在高效催化剂作用下,可以将CO2催化转化为高附加值化学品,对CO2减排和资源化具有重要意义。对近年来CO2和胺/H2(氢硅烷)催化转化为N-甲酰胺和N-甲基胺的催化剂体系、反应工艺和反应机理等方面的研究进展进行归纳、总结和评述,并对研究工作的发展趋势进行展望。
In the process industrialization,a large amount of greenhouse gas CO2 is discharged into the atmosphere,and its impact on the atmospheric environment is getting more and more serious. However,CO2 is also a cheap carbon source. Under the action of high-efficiency catalysts,CO2 can be catalytically converted into high-added-value chemicals,which is of great significance for CO2 emission reduction and resource utilization.This article reviews the latest researches in catalytic conversion of CO2 and amine/H2 (hydrosilane) to N-formamide and N-methylamine in terms of catalyst system,reaction technology and reaction mechanism,as well as the prospects.
受能源需求推动, 化石燃料排放量增加, 大气中CO2含量不断升高。据能源部统计, 2019年底, 全球化石燃料产生的CO2排放量达3.68× 1011 t, 高于2018年3.657× 1011 t[1]。作为一种温室气体, CO2超量给环境带来一系列问题, 如全球气候变暖、冰川融化和海平面上升等[2, 3]。值得注意, CO2是一种绿色且廉价的碳资源, 可以作为合成有机化学品的C1源[4], CO2转化为有价值产品的途径主要有矿物碳酸化、化学转化和生物转化。矿物碳酸化可成为建筑业产品, 如混凝土; 而化学和生物转化都可以产生有价值的工业化学品, 如甲醇或甲酸, 以及燃料作为产品[5]。就规模而言, 只有少数工业过程已实现CO2利用, 如尿素[6]、水杨酸[7]和碳酸盐[8]的生产。此外, 还报道了CO2催化转化为甲醇、甲酸或其衍生物。CO2也可以转化成环状碳酸酯[9]和羧酸等具有高附加值的产品。
近年来, CO2的催化转化引起越来越多研究人员的注意。通过化学方法固定CO2, 在还原剂H2或有机硅烷的存在下可以转化为甲酸[10]; 在Fe基、Co基、双金属和复合催化剂等高效催化剂作用下, CO2为碳源、H2为还原剂, 可以将CO2转化为C2H4和C3H6等低碳烯烃[11]; CO2为碳源, H2或有机硅烷为还原剂, 使用Pd基、Cu基催化剂可以生成甲醇[12]高附加值产物; CO2还可以和H2或有机硅烷及胺进行N-甲酰化和N-甲基化反应生成精细化学品中间体[13], 适用的催化剂体系主要是Au、Pd、Ru和Pt等贵金属催化剂, Cu、Ni、Co、Mn和Zn等非贵金属催化剂和离子液体、N杂环卡宾化合物等非金属催化剂。三种催化剂各有优缺点, 贵金属催化剂催化活性和选择性好, 但成本高, 反应条件苛刻; 非贵金属催化剂和非金属催化剂价格低廉丰富, 但催化活性不如贵金属催化剂, 且不易回收。因此, 探索具有高活性和高选择性的CO2还原的新型催化剂具有重要意义。
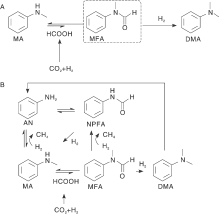
许多酰胺类化合物具有工业用途, 在有机合成和药物化合物合成中具有广泛应用。此外, N, N-二甲基甲酰胺作为重要工业溶剂以及用于合成转化的多功能试剂。传统上, 通常使用危险、有毒性、原子经济低的乙酸甲酸酐、甲酸等羰基化合物作为羰基源引入胺中进行生产[14]。与传统合成路线相比, CO2的还原性官能化为甲酰胺的合成提供了潜在的环境友好的替代方案。甲基取代胺被广泛用作制造药品、农用化学品、染料或用作溶剂的关键中间体[15]。工业中胺甲基化最常用的方法是使用有毒溶剂甲醛作为C1源[16], 因此, 需要研究安全且可持续碳源和高活性催化剂投入应用。
本文对近年CO2作为C1源、H2或有机硅烷作为还原剂催化转化胺N-甲酰化或N-甲基化反应生产高附加值产品的催化剂体系、性能、工艺条件和催化反应机理等研究进展进行归纳及展望。
Au催化剂具有高活性和高选择性以及极高稳定性, 常被作为非均相催化剂研究。均相催化剂在此领域已取得显著进展, 但对非均相催化剂的报道并不多, 这是由于非均相催化剂具有低周转率TOF(小于3.3 h-1), 以及要想实现高的产物收率就需要延长反应时间超24 h, 极大限制了非均相催化剂的应用。Du X L等[17]利用Au催化剂的优异性能负载于Al2O3上得到非均相催化剂Au/Al2O3-VS, 140 ℃和7 h完成了苯胺的N-甲基化反应, 苯胺完全转化, 产率达92%, 得到目的产物N, N-二甲基苯胺。测试了Au原子表面的平均周转率, 是有史以来报道CO2与胺/H2反应的最高值, 达45 h-1。此外, 研究了负载在Al2O3上其他贵金属(Pt、Ir和Rh)的活性, 发现Au活性远高于其他贵金属。还研究了带有不同类型基团芳香胺的活性, 发现具有给电子基团的苯胺活性基本不变, 而带有吸电子基团的苯胺反应活性降低。Au粒径大小直接影响催化剂活性。Tang G等[18]利用沉积-沉淀法制备Au/γ -Al2O3催化剂, 重点研究老化温度、老化时间、尿素与Au物质的量比对Au平均尺寸的影响。发现随着老化温度由20 ℃增到80 ℃, Au粒径由8.3 nm缩小到1.8 nm; 老化时间6 h比1 h的粒径尺寸小; 尿素控制Au沉淀溶液的pH值, 所以加入量过多过少都不利于Au的沉积, 当尿素和Au物质的量比为200时, Au大部分负载于Al2O3上。这样负载的Au最小粒径可以到1.8 nm, Au尺寸越小, 反应产率越高。
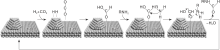
目前研究的大部分催化剂仅适用于一些简单的胺, 而对于含有官能化基团的胺来说, 反应选择性低仍是不可避免的问题。Takato K等[19]发现, 催化剂Au/TiO2表现出优异的活性和选择性, 可以对各种胺进行N-甲酰化反应, 尤其是带有可还原官能团的胺, 反应结束后, 可还原基团完全保留, 表现出高选择性。对带有烯烃基团的脂肪胺, 当Au颗粒平均粒径4.2 nm时, 催化活性最高, 收率达99%。与之形成鲜明对比的是, Pd、Ru、Pt、Rh、Ag和Cu金属颗粒的选择性和活性都很差; 载体种类方面, TiO2最有效。还从反应混合物中发现了甲酸, 推测Au纳米粒子攻击CO2氢化成甲酸, TiO2吸附的胺与甲酸反应, 使得官能化胺可以高选择性N-甲酰化(见图1)。
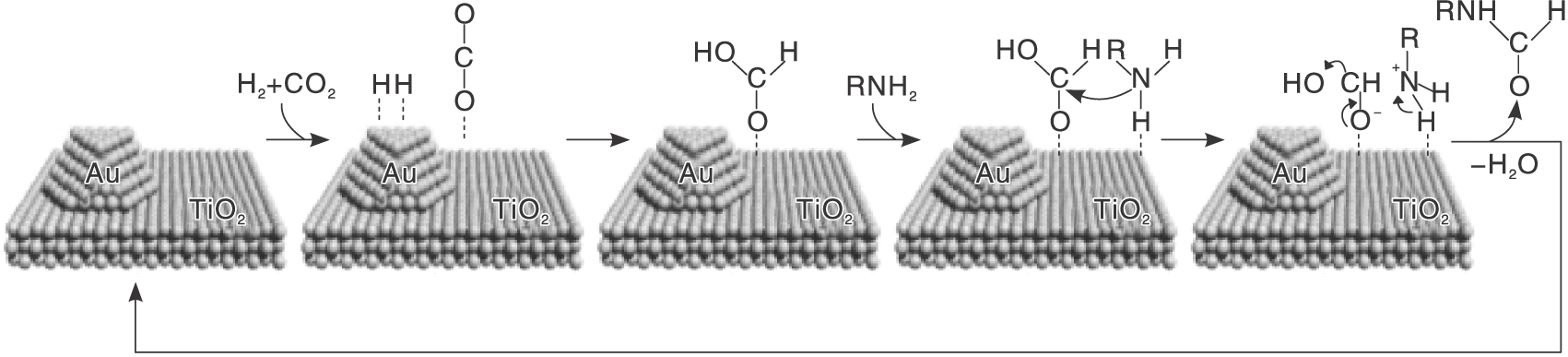 | 图1 Au/TiO2催化CO2与胺/H2的N甲酰化反应路径[19]Figure 1 Proposed reaction path for Au/TiO2-catalyzed N-formylation of amines with CO2 and H2 |
随着对CO2/H2与胺反应的深入研究, 以及对以往催化剂特性的分析, 研究人员发现既具有酸性位点又具有碱性位点的催化剂似乎对该反应更具活性。而MOF作为一种新发展起来的材料, 具有可调的孔径和配位不饱和金属中心, 引起研究人员的广泛注意。通过热回流法合成的MOF-808(Zr)[20], 具有不饱和Zr金属配位, 负载不同含量的Au颗粒, 发现使用质量分数3.0%的Au/MOF-808(Zr)催化剂催化苯胺反应, 得到选择性80.9%的N-甲基苯胺, 选择性19.1%的N, N-二甲基苯胺。通过对甲酸、CO2和甲醇作对比实验, 研究该反应的初步机理, CO2和苯胺/H2反应过程中, CO2和H2很可能反应生成甲酸, 甲酸随后与苯胺发生N-甲酰化或N-甲基化反应。同一研究组通过溶剂热合法制备MIL-101(Cr)[21], 并负载Au纳米颗粒合成Au/MIL-101(Cr), 催化CO2与苯胺/ H2反应, 显示出良好的催化效果和重复使用性能。
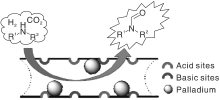
Pd作为贵金属活性组分, 已被证明是氢化反应中真正的活性位点。Zhang Y J等[22]选择易于功能化的碳材料, 利用羟基亲水性质, 在碳材料表面官能化调节Pd/C催化剂活性。添加碱50%KOH做助催化剂进一步提高催化剂性能, N-甲酰基哌啶产率达99%。研究进一步发现, 催化剂对脂肪伯胺具有良好的催化活性。通过共沉淀法制备载体MgAl LDH(LDH是一类合成阴离子黏土), 负载Pd贵金属。研究重点着重于溶剂与CO2与胺/H2的N-甲酰化反应之间的联系。实验测试了甲醇、己烷和四氢呋喃(THF)溶剂对反应的影响, 发现己烷和THF的副产物远高于甲醇, 原因是甲醇存在时, 为反应提供了HCOOCH3作为中间体的反应途径。由此可见, 溶剂甲醇在N-甲酰化反应中起重要作用。非均相催化剂在使用过程中存在反应条件相对苛刻, 且载体制备复杂。Dai X C等[23]发现了一种载体PAL(一种天然可用的一维纳米级水合镁铝硅酸盐黏土矿物), 具有丰富的通道及酸碱性位点, 利用该载体负载Pd得到的非均相催化剂, 在相对温和条件下对伯胺和仲胺具有良好的催化活性。推测Pd/PAL优异的催化性能得益于PAL中固有的酸和碱性位点与金属Pd的协同作用(见图2)。Cui X等[24]根据甲醇和胺合成N-甲基胺的思路设计了一种将氧化铜和过渡金属氧化物结合的活性催化剂, Pd作为该反应的活性组分负载于载体上得到Pd/CuZrOx, 还将Al、Zr、Zn、Fe、Mg和Ni金属氧化物与Cu和Zr结合以期望优化催化剂性能, 结果并不理想, 甚至催化活性反而下降, 认为Pd/CuZrOx具有优异活性得益于高比表面积和良好的介孔结构以及Pd、Cu和Zr之间的协同作用, 并提出N-烷基甲酰胺可能是加氢合成N-甲基胺的中间体。
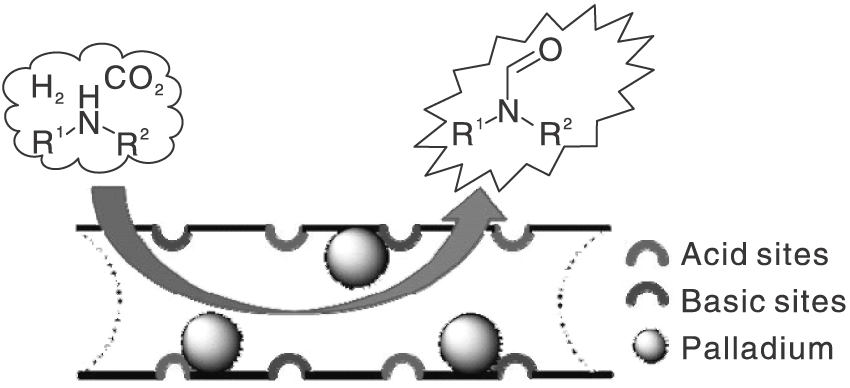 | 图2 Pd/PAL催化剂上胺与CO2和H2的N-甲酰化[23]Figure 2 N-formylation of amine with CO2 and H2 catalyzed by Pd/PAL catalyst |
Pd和Au双金属催化剂作为非均相催化剂经常用于CO2还原, 因而是目前研究最广泛的双金属催化体系。Ju P P等[25]报道了Pd-Au负载型双金属催化剂, 载体为聚苯胺官能化碳纳米管, 用于吡咯烷的N-甲酰化反应, 相应的N-甲酰基产物在125 ℃产率为98.3%。相比较单一金属负载型催化剂, Pd催化剂得到酰基化产物的产率为50%, 而Au催化剂仅为3.9%。显而易见, 双金属催化剂Pd-Au/PANI-CNT活性更显著。作者提出酰基化反应主要发生在Pd原子或Pd原子和Au原子的界面, 因此Pd-Au/PANI-CNT的酰基化性能增强主要与Au与Pd合金化引起的双金属Pd-Au纳米粒子的电子或电子特性的变化有关。
关于CO2和H2进行N-甲基化反应的关键, 大量的研究工作指向探索适合的活性催化剂活化CO2, 使非均相反应转化为均相反应。Su X等[26]根据前人研究提出的反应机理设计了一款有效的非均相催化剂PdGa/TiO2, 通过表征手段证实PdGa双金属纳米颗粒分散负载在TiO2载体上得到PdGa/TiO2, 用于N-甲基苯胺的N-甲基化反应, 反应显示98%转化率和94%选择性, 是非常理想的催化结果。研究表明, 在载体TiO2上, Pd可以与Ga相互作用转变成缺电子Pd 将CO2活化成甲酸。通过对比试验发现, 在甲酸存在下, 转化率更高, 而产生这一良好催化性能正是由于制备的高效催化剂PdGa/TiO2可以活化CO2, 并将其转化成甲酸, 这是CO2/ H2进行甲基化反应至关重要的一步。根据大多数研究人员提出的反应机理:CO2与H2反应, 在催化剂表面形成甲酸, 甲酸盐类与胺进一步反应生成甲酰胺类中间体, 接着被H2快速还原成N-甲基化产物。Lin W W等[27]发现PdZn合金在稳定甲酸盐中间体方面起重要作用, 制备的一系列Pd-ZnO/TiO2、Pd/TiO2和Pd/ZnO催化剂, 比较其催化活性, Pd-ZnO/TiO2对该反应表现出更高催化性能, 选择性更高, 选择性接近100%。ZnO的引入可以促进CO2与H2形成甲酸, 还可以阻断由于N——C键断裂产生副产物的路径, 对选择性有非常积极的影响, 而TiO2载体的加入改善了PdZn合金的分散性。通过分析反应曲线, 发现甲酸的形成与PdZn合金的数量成正比, 即PdZn合金是该催化剂对反应表现出高催化活性的原因。作者还探究了具有取代基团的苯胺底物的反应活性, 具有供电子基团的苯胺转化率和选择性都有略微提高, 具有吸电子基的苯胺转化率下降严重, 而且反应活性不仅与反应底物的电子密度有关, 还与取代基的位置有关(图3)。
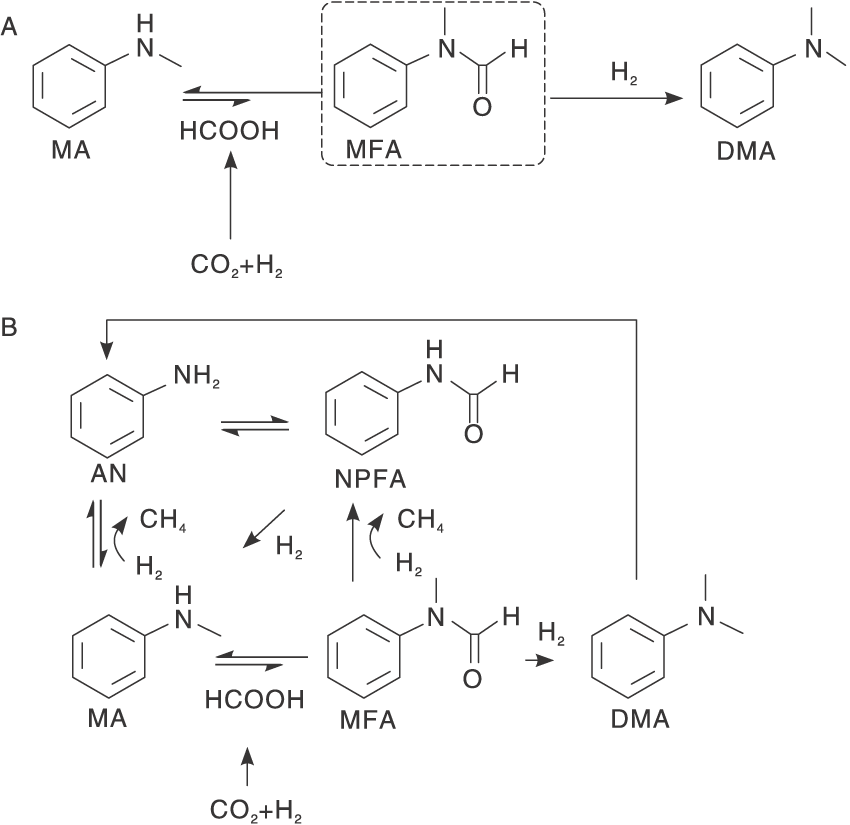 | 图3 MA与CO2和H2的(A)PdZn和(B)Pd催化的N甲基化反应途径[27]Figure 3 Reaction paths for (A) PdZn and (B) Pd-catalyzed N-methylation of MA with CO2 and H2 |
Pt在非均相催化剂中也有一定应用。Toyao T等[28]利用顺序浸渍法制备Pt和MoOx负载的TiO2催化剂, 在无溶剂下催化氨气及氨气替代物, 得到收率65%的三甲胺。测试了过渡金属氧化物负载的质量分数对催化剂活性的影响, 发现质量分数5%过渡金属氧化物负载的催化剂活性最高; 测试负载各种金属(Pt, Pd, Re, Ir, Rh, Ru, Ni, Co, Cu)的催化活性, 最终得出Pt-MoOx/TiO2催化氨气转化为三甲胺的合成产率最高, 而MoOx/TiO2表现出零产率, 表明Pt的存在对该反应的进行至关重要。作者还提出对于氨气转化三甲胺反应过程中可能涉及到甲酸的形成。
贵金属催化剂具有良好的活性和高选择性, 但单负载贵金属催化剂在高温反应后易出现贵金属成分流失、团聚和烧结等问题, 通过双金属改性引入过渡金属和其他种类的金属或使用既具有碱性活性位点, 又具有酸性活性位点的载体, 就可以优化活性组分的活性, 从而在提高催化剂活性的同时, 提高N-甲酰化产物或N-甲基化产物的选择性和收率。
与贵金属催化剂相比, 地球富含过渡金属, 特别是Fe、Co、Ni、Mg、Mn、Zn和Cu是用于大规模生产过程的有效催化剂, 许多研究者致力于研究活性高、反应条件温和的非贵金属催化剂。
铜基催化剂价格便宜, 用途广泛, 在CO2催化方面表现出优异活性。Liu J等[29]比较了Al2O3、ZnO金属氧化物负载Cu对CO2/H2与胺反应活性的高低, 发现Cu/ZnO的活性优于Cu/Al2O3, 而且Cu/ZnO可以在无溶剂条件下催化CO2、H2和二甲胺反应生成二甲基甲酰胺(DMF)。还提出由Cu/ZnO催化的CO2、H2和二甲胺合成DMF的反应机理:铜可以激活氢, 并且在铜表面产生甲酸盐物质, 生成的甲酸盐氢化为甲酸并与二甲胺反应形成二甲胺甲酸盐, 脱水形成DMF。合适的金属Cu与Zn物质的量比为3∶ 2, 使Cu和ZnO的协同效应发挥到最优。
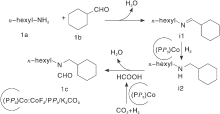
碱金属络合物也被常用于研究活化CO2。碱金属/配体催化剂活性受金属、配体取代基和二膦配体的螯合咬合角大小影响。作者筛选17种过渡金属盐和几种不同类型的配体得到Fe(Ⅱ )和Ni(Ⅱ )dmpe络合物[30], 其在胺存在下氢化CO2的催化活性很高, 转换数高达18 000, 惟一不足是反应压力高达6 MPa。还研究了在无溶剂条件下用吗啉和2-乙基己胺催化氢化CO2, 对比加入溶剂二甲基亚砜(DMSO)对于CO2氢化, 溶剂DMSO起重要作用, 使得转化数(TON)值高于未添加溶剂时观察到的TON值。Jayarathne U等[31]研究PNP钳形配体支持的羰基氢化铁, 测试3种不同类型胺底物苯胺、二苯胺和吗啉的反应活性, 仅吗啉产生了N-甲酰化反应, 苯胺转化率非常低, 因为芳香胺的碱性和亲核性相对较弱。带有给电子取代基的胺底物更有利于N-甲酰化, 因此可以增强底物碱性或改善亲核性。Ke Z等[32]报道了一种高选择性合成非对称N, N-二取代甲酰胺的新路径, 通过伯胺和醛与CO2/H2在CoF2、P(CH2CH2PPh2)3和 K2CO3组成的钴基催化体系上还原偶联而得到, 该络合物具有良好的官能团耐性和广泛的底物范围。反应溶剂对催化活性也有显著影响, 非极性溶剂抑制反应发生, 高极性非质子溶剂如EtOH、DMSO和DMF, 为反应提供不错收率。还研究了反应机理:通过醛和催化剂作用, 将伯胺氢化形成仲胺, 在Co催化剂作用下CO2氢化脱水产生HCOOH, 仲胺与HCOOH最终生产N-甲酰化产物(图4)。
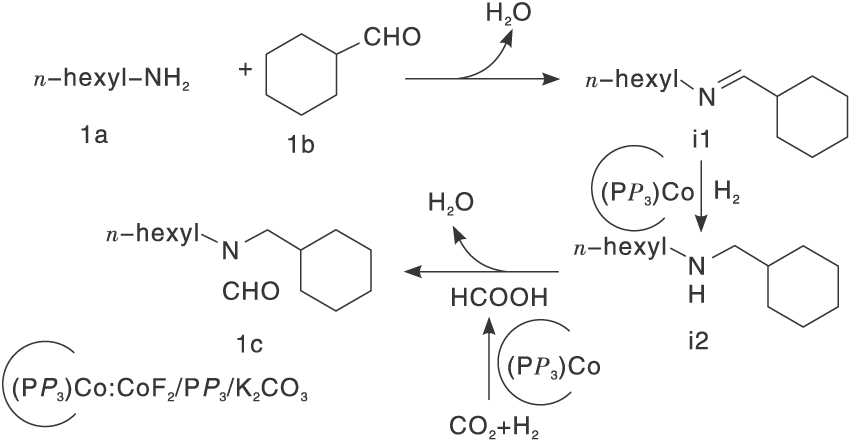 | 图4 伯胺、醛与CO2/H2在CoF2 P(CH2CH2PPh2)3和K2CO3组成的催化体系的机理研究[32]Figure 4 Plausible reaction mechanism for reductive coupling of primary amine and aldehyde with CO2 and H2 |
Daw P等[33]设计Co-PNP钳形复合物Co-PNP, 可以在相对温和条件下对广泛范围的伯胺和仲胺进行N-甲酰化, 首次报道使用碱金属络合物催化CO2和胺/H2进行N-甲酰化。NaHBEt3和tBuOK在Co-PNP产生的催化活性物质中起着不可替代的作用。作者还提出了反应机理:Co-PNP在两个碱物质作用下产生配位不饱和中间体, 并在H2和CO2气氛下形成Co氢化物和η 1-形式复合物, 胺对形式复合物直接亲核攻击, 随后从金属中心释放H2O形成N-甲酰化产物。
Toyao T等[34]通过简单的浸渍方法制备多相催化剂Re/TiO2用于氢化反应展示出优异的选择性。这是由于与其他催化剂相比, Re对羰基基团的亲和性高于苯环, 在加氢反应不会产生脱芳烃副产物, 因此表现出高选择性。将该催化剂用于胺的N-甲基化也产生了明显的催化效果, 在
非贵金属催化剂的生产成本相较贵金属催化剂低很多, 但随之而来的是低活性和低选择性, 还原剂通常使用还原效果更好的硅氢烷, 因此开发高活性和高选择性的N-甲基化和N-甲酰化催化剂是未来工业化发展的需求。CO2/H2与胺催化转化为N-甲酰胺或N-甲基胺类化合物如表1所示。
| 表1 CO2/H2与胺催化转化为N-甲酰胺或N-甲基胺类化合物 Table 1 Proposed reaction path for Au/TiO2-catalyzed N-formylation of amines with CO2 and H2 |
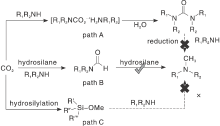
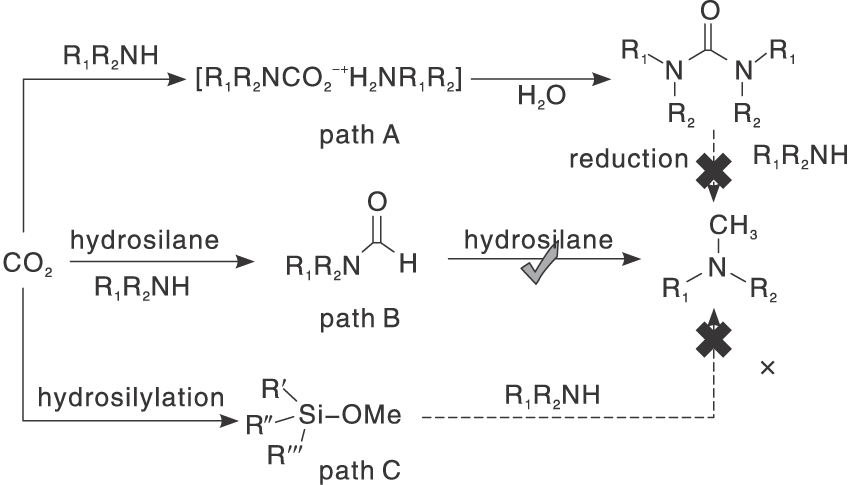
氢硅烷作为一类有效的还原剂, 由于其Si— H键比H— H键更弱且更具极性, 所以具有轻度还原电位的极性Si— H键在动力学上比H— H键具有更高的反应活性, 而且反应条件更温和。此外, 产物中形成的强Si— O键提供了额外的热力学驱动力。
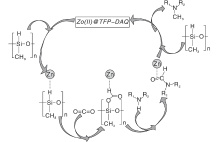
丰富且价格低廉的过渡金属由于空的d轨道的存在, 常被作为非贵金属催化剂与配体形成配合物。Jacquet O等[35]采用工业锌盐和IPr作为辅助配体形成IPrZnCl2配合物, 首次报道CO2和氢硅烷作为还原剂与胺的N-甲基化反应。探索该新型催化剂对不同有机硅烷的活性, 实验表明, PhSiH3在N-甲基苯胺的甲基化反应中比Ph2SiH2、Ph3SiH、Et3SiH和(EtO)3SiH更具反应性。目前发现金属配合物催化CO2和硅烷对N— H键甲酰化和甲基化理论研究很少。Li W Y等[36]拓展了前者工作研究反应机理以及研究了NHC(IPr)配体在催化反应中的作用, 通过详细地DFT计算分析, 得出总体反应机理概况为两个阶段:N-甲酰苯胺的形成和N-甲酰苯胺向N, N-二甲基苯胺产品的还原。IPr配体在反应中主要起两个作用:(1) NHC配体与锌中心的配位阻止N-甲酰苯胺物种与锌中心的配位, 从而避免N-甲酰苯胺中羰基氧原子因配位造成亲核性降低而影响反应活性。(2) IPr配体降低了锌中心的路易斯酸性, 有利于保持Si— H键的活性。Hota P K等[37]发现aNHC比正常NHC更具亲核性。乙腈作为探索反应的溶剂, 通入低于1 MPa的CO2压力, 使用物质的量浓度5%的aNHC催化酰胺甲基化, 可以在环境温度下得到产率74%的N-甲基化产物。共价有机骨架(COF)是新发展的材料, 是共价排列的晶体多孔二维(2D)材料, 具有高BET表面积和热稳定性的低序低密度框。Sarkar P等[38]在TFP-DAQCOF载体上负载锌催化剂, COF材料其优异的中孔结构及高表面积可以在温和条件下进行反应, 同时可以通过改变溶剂的类型控制底物发生N-甲基化和N-甲酰化反应, 其中使用乙腈、1, 4-二恶烷可以得到高产率的N-甲基化产物, 使用THF溶剂可以收获高收率的N-甲酰化产物, 而在甲苯和DMF存在下, 不发生任何反应。作者还提出了可能的反应路径:Zn金属活化氢硅氧烷(PMHS)的Si— H键, 然后将CO2插入Si— H键中, 还原羰基以获得N-甲基化胺产物(图5)。
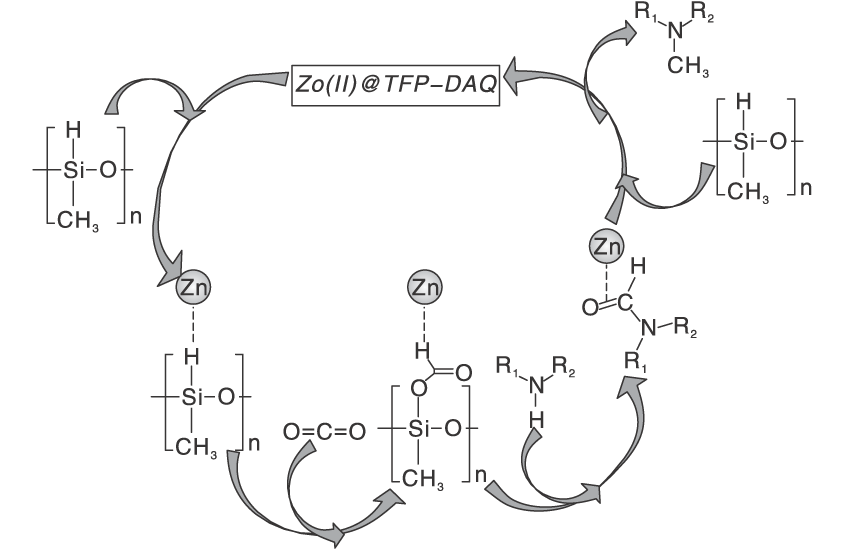 | 图5 Zn(Ⅱ )@TFP-DAQ COF催化剂上胺的N-甲基化的拟议机理途径[38]Figure 5 Proposed mechanistic path for N-methylation of amine over Zn(Ⅱ )@TFP-DAQ COF catalyst |
即使这些过渡金属催化剂对反应的活性很高, 但仍存在一定的缺点, 如需要使用含膦配体、在反应中加入极性溶剂如CH3CN, 四氢呋喃(THF), CH2Cl2等来提高催化活性。Luo R C等[39]发现高极性有机溶剂可以通过溶剂化和极化激活胺中的N— H键, 使用锌酞菁(ZnPc)和DMF极性溶剂催化胺的N-甲酰化反应, 显示出高稳定性和催化活性。反应可以在很低的温度25 ℃下和0.1 MPa的CO2压力下反应产物还可以达到较好的收率。极好的反应活性和 ZnPc与DMF之间的协同作用密切相关, 推测ZnPc和DMF可能促进氢原子从含氢硅烷到CO2的C=O键的亲核加成, 这是N-甲酰化反应的关键步骤。
Lucero G S等[40]首次证明了Ni金属对CO2/脂肪胺的N-甲基化反应有效。使用苯胺作为反应底物, 比较镍基催化剂[(dippe)Ni(μ -H)]2和市售Ni(COD)2/dcype催化剂的N-甲基化性能, 发现这两种催化剂在反应后产生4种产物, 但主要产物还是N-甲基苯胺。Huang Z J等[41]新开发的催化剂[Mn2(CO)8(L1)2]是一个对称体, 由2个Mn中心和2个L1分子组成, 是第一个双核锰配合物可以对芳基伯胺进行选择性双N-甲酰化的催化剂。研究表明, 胺的N-甲酰化反应可能主要分为两个阶段:(1) CO2与硅氢加成反应形成甲硅烷基甲酸酯, 与胺形成甲酰化中间体, (2) 甲酰胺还原为甲基化产物。
碱金属具有非常强的金属性。Fang C等[42]比较了Li2CO3、Na2CO3、K2CO3、Rb2CO3和Cs2CO3碱金属盐的催化活性, Li2CO3和Na2CO3对反应没有活性, 随着阳离子尺寸的增加, 活性提高, 产物产率也得到进一步提高, Cs2CO3活性最高, 将这一现象归结为铯效应和碳酸盐溶解度增加以及亲核性增强。测试Cs2CO3有广泛的底物范围, 有20种之多, 且都有很好的选择性和转化率。通过控制反应温度、时间和硅烷剂量可以实现对N-甲基苯胺选择性的N-甲酰化或N-甲基化, 在室温下, 加入少量Ph2SiH2, 反应12 h, N-甲基苯胺主要发生N-甲酰化反应, 收率达94%; 反应温度80 ℃, 加入较多Ph2SiH2, 反应24 h, 主要得到N-甲基化产物, 产物收率达92%, 而且相同的Si— H键, Ph2SiH2比Ph3SiH和(EtO)3SiH产率高。相比于过渡金属催化剂及N-杂环卡宾等具有配体的复杂催化结构, 碱金属Cs2CO3具有简明的催化结构, Cs2CO3操作制备简单, 最重要的是可以高选择性催化胺与CO2和硅烷进行甲酰化/甲基化以生产酰胺/甲基胺类产物(图6)。
 | 图6 Cs2CO3催化胺与CO2和氢硅烷甲基化的潜在途径[42]Figure 6 Potential pathways for methylation of amineswith CO2 and hydrosilane catalyzed by Cs2CO3 |
无过渡金属催化主要取决于催化剂与还原剂之间的酸碱相互作用, 具有酸性或碱性性质的催化剂往往是激活Si— H的关键。Jiang X等[43]开发的基于金属钠的芳基硼酸酯静电催化剂用于胺的N-甲酰化, 探索不同含硼化合物作为胺N-甲酰化催化剂的反应活性, 发现芳香族硼酸对于酰胺的氢化硅烷化还原反应没有效果, 带有吡啶基的硼酸活性也很低, 其他芳香族杂环硼酸和硼酸酯同样没有活性, 而三羟基芳基硼酸钠具有可靠的催化活性, 主要是由于其具有酸性质子和碱性氧原子, 可以有效地激活反应底物的Si— H键。作者还探索了使用不同金属钠碱制备芳族硼酸钠, 发现金属钠碱不同时, 反应活性也不同, MeONa、EtONa、tBuONa或PhONa作为对位碱时, N-甲基甲酰苯胺收率在34%~68%范围内不等, 用EtONa制备的芳族硼酸钠在相同反应条件下具有更好的反应性, 提高反应温度, 反应活性更好, 收率更高。对带有官能团的胺底物N-甲酰化反应具有良好的反应性和选择性, 对反应难度较大的吡啶基胺的甲酰化反应同样有效。因此, 碱金属催化剂对胺与CO2催化还原转化提出一种新的思路。
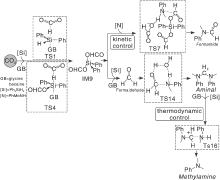
天然存在的化合物由于具有绿色、可再生等特点已经被作为非金属催化剂用于胺与CO2和有机硅烷的N-C反应中, 同时也获得越来越多的关注。甘氨酸甜菜碱[44](GB)是一种天然的具有阴阳离子结构的季铵盐, 可以用作碱性催化剂使用, 发现通过改变反应温度和反应物物质的量比可以控制反应类型和产物类型, 室温下, n(胺)∶ n(PhSiH3)∶ n(CO2)=1∶ 2∶ 10时, 主要发生N-甲酰化反应; 当温度升高到100 ℃时, n(胺)∶ n(PhSiH3)∶ n(CO2)=1∶ 2∶ 2时, 生成N-甲基胺主产物; 同样温度下, n(胺)∶ n(PhSiH3)∶ n(CO2=1∶ 4∶ 6时, 主要产物变为N, N-二甲基胺。与甘氨酸甜菜碱类似, 卵磷脂[45]具有阴阳离子结构而且比甘氨酸甜菜碱的来源更广泛。当使物质的量浓度5%卵磷脂作为催化剂时, 反应在相同反应条件下有效进行, N-甲基苯胺完全转化, 得到N-甲基甲酰苯胺产率97%, 比甘氨酸甜菜碱活性略高。还检测了其他含氢硅烷(即Ph2SiH2、Ph3SiH)的活性, 但是他们都不能提供甲酰化产物。对于伯胺, 由于两个N— H键具有相同的反应性, 通常会产生两种胺的产物单-甲酰化和双-甲酰化产物, 但在该催化体系中并没有双甲酰化产物, 这是因为卵磷脂的空间位阻较大, 所以卵磷脂可以作为伯胺选择性甲酰化得到单甲酰化产物的高选择性有机催化剂。Yue Z等[46]提出反应中产生的活性中间体甲硅烷基甲酸酯, 是反应速率的决定步骤, 这一点与上述研究人员推测的机理基本一致。在天然化合物催化剂作用下, Ph2SiH2多次连续攻击使得CO2被还原, 生产稳定的活性中间体甲硅烷基甲酸酯(IM9)。通过控制温度、反应物之间的物质的量比和反应时间, 就可以控制N-甲酰化/N-甲基化的选择性(图7)。
 | 图7 GB催化的N-甲基苯胺和Ph2SiH2还原CO2生成甲酰胺、氨基和甲胺的还原功能[46]Figure 7 GB catalyzed reduction functionalization of CO2 with N-methylaniline and Ph2SiH2 to produce formamide, amine, and methylamine |
Mu Z J等[47]合成两性离子COFs材料后, 通过孔隙表面工程方法将甜菜碱基团引入COFs通道壁。该非均相催化剂的高稳定性、不溶性、可调催化位点以及来自其多孔结构的一维通道的良好质量传递, 表现出良好的催化活性。其为开发具有突出性能的有机催化剂提供了一种新方法。
Hao L D等[48]用咪唑鎓阳离子和ILs阴离子制备咪唑离子液体(ILs), 对催化N-甲酰化反应具有极好的协同效应。([BMIm]Cl)对该反应非常有效, 在室温反应5 h, 得到N-甲基甲酰胺产率93%。比较具有相同阳离子的其他4个IL(即[BMIm]+)的反应活性, [BMIm]Br和[BMIm]NO3均有活性, 而[BMIm]PF6和[BMIm]BF4对反应无效。结果表明, IL的阴离子对IL的活性具有显着影响。通常, 溶剂在促进质量传递和增强催化剂的亲核性方面起促进作用。Zhao W F等[49]制备乙酰胆碱-羧酸盐生物离子液体(ACH-AAIL), 通过控制反应温度, 反应溶剂可以选择性生成甲酰胺或甲胺产物。
Shen Q等[50]制备无金属催化剂氮掺杂石墨烯纳米片(NG)。鳞片状石墨比表面积7.76 m2· g-1, 通过尿素(H2NCONH2)、碳酸钾(K2CO3)和高温处理得到NG表面积升高到684.32 m2· g-1, 形成无序微晶排列的微孔和中孔结构。催化苯胺N-甲酰化, 苯胺可以完全转化, 在60 ℃下N-甲酰化产物收率达97%。Das S等[51]从维生素B1衍生出高效且廉价的基于噻唑鎓碳烯基的催化剂, 使用聚甲基氢硅氧烷(PMHS)作为还原剂, 将含氮药物Cinacalcet的N-甲基化, 可增强其活性。图2为CO2/氢硅烷与胺催化转化为N甲酰胺或N-甲基胺类化合物。
| 表2 CO2/氢硅烷与胺催化转化为N甲酰胺或N-甲基胺类化合物 Table 2 Catalytic conversion of CO2/organosilane and amine into N-formamide or N-methylamine compounds |
酰胺类化合物和甲基取代的胺类化合物在药品和农用化学品等方面均有广泛应用。以廉价CO2作为C1源对胺类化合物进行N-甲酰化/N-甲基化反应, 不仅降低了反应成本, 提高了胺类化合物的附加值, 还为CO2回收利用提供了一条新路径。对于CO2与胺/H2(氢硅烷)催化转化成N-甲酰胺或N-甲基胺化合物的研究, 催化剂的选择和设计是关键。研究人员多采用具有酸碱活性位点的氧化物和MOF等材料负载贵金属或非贵金属作为反应的催化剂, 以提高对CO2、H2和胺类化合物的吸附, 促进反应在催化剂表面进行, 从而获得高转化率和选择性。目前研究人员普遍认为该反应机理是:对于H2作还原剂的反应, 金属催化剂攻击CO2与H2生成甲酸, 载体吸附胺类化合物与甲酸中间体反应生成N-甲酰化产物, 进一步加氢生成N-甲基化产物, 甲酸是中间体; 对于有机硅烷作还原剂的反应, 硅酸酯是反应中间体。
随着科学技术的进步和研究手段的不断提高, 对于催化剂的设计和反应机理的研究会越来越深入。设计价格低廉且丰富的催化剂, 在控制反应进行方向的同时, 提高催化剂选择性和活性, 延长催化剂循环利用率, 尽可能选择环境友好型还原剂如H2, 这都将是研究人员在CO2与胺类化合物反应中需要努力的方向。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|