{kind=link}
甲烷活化与催化转化过程中的几个关键问题及对策
[肖晓敏, 曹学雨, 吴新宇, 耿豪杰, 於思瑜*  , 刘社田
, 刘社田* ]
, 刘社田]
|
|


作者简介:肖晓敏,1995年生,女,在读硕士研究生,研究方向为工业催化。
现有的由甲烷直接转化生产大宗化学品过程,包括氧化偶联制乙烯、选择性氧化制甲醇和脱氢低聚制芳烃等,依然停留在实验室研究阶段,无法达到工业化要求。其原因在于单程目的产物收率低,能耗高,无法和现有生产过程竞争。而其技术障碍则与甲烷C—H键活化条件苛刻、产物不稳定、催化剂选择性和稳定性差等因素相关。本文主要分析了实现甲烷活化和高效直接转化过程中的一些关键技术问题,主要包括临氧转化过程中的选择性氧化、脱氢低聚过程中的热力学平衡限制及催化剂稳定性等,并对其可能的解决途径进行了探讨。
Existing technologies for direct conversion of methane to bulk chemicals,including oxidative coupling to ethylene,selective oxidation to methanol and dehydro-oligomerization to benzene,are still in their bench-scale phase and far away from commercialization,due to low single-pass yield of target products and high energy consumption. The technical obstacles are related to factors such as harsh condition for methane C—H bond activation,poor stability of the target products,and poor selectivity and stability of the catalysts. This paper analyzes some key problems with the process of methane activation and high-efficient direct conversion,including selective oxidation oxidative conversion,thermodynamic equilibrium limitation in dehydrogenation and oligomerization,and stability of the catalysts. Possible solutions to these problems are discussed.
甲烷是最简单的有机物, 也是含氢量最大的烃, 主要以天然气、页岩气、煤层气和甲烷水合物的形式存在。由于其在地壳中蕴藏量丰富, 是短期内可以有效取代煤炭、石油的清洁能源和化工原料。甲烷作为化工原料可以生产有机材料、液体燃料及其它化学品。由于甲烷分子中氢含量远高于大多数化学品, 脱氢是甲烷转化的关键步骤, 甲烷转化为化学品可以副产大量的氢气。因此, 甲烷也是氢能产业发展中值得特别重视的能源资源。通过提高甲烷的使用量, 可以有效地降低二氧化碳的排放, 有利于达成国家制定的碳达峰和碳平衡的能源战略目标。本文主要分析了实现甲烷活化和高效直接转化过程中的一些关键技术问题, 主要包括临氧转化过程中的选择性氧化、脱氢低聚过程中的热力学平衡限制及催化剂稳定性等, 并对其可能的解决途径进行了探讨。
甲烷分子拥有特殊的正四面体结构, 其C— H键能达到439.4 kJ· mol-1[1]。这使得甲烷在常规条件下难以活化, 通常需要在强酸强碱或高温条件下进行, 且生成的产物活性或反应性能远高于甲烷, 易进一步转化, 导致目的产物选择性难以控制。这是从甲烷合成高附加值化学品所面临的核心挑战。
通常甲烷分子的活化需要催化剂的作用, 活化机理可以分为以下三类:(1)C— H键与催化剂形成金属-碳σ 键的中间体或最终产物; (2)C— H键仅与催化剂作用, 金属-碳σ 键不在任何阶段直接产生; (3)催化剂首先与另一种反应物形成活性中间物, 然后攻击甲烷分子使C— H键活化。有关甲烷活化机理方面的论述可见文献[2, 3]。这里主要讨论在工业上有重大应用价值和前景的甲烷转化过程。一般来讲, 甲烷的转化途径可以分为间接转化与直接转化两种。
甲烷的间接转化过程是将甲烷首先转化为不同CO/H2比例的合成气(CO+H2), 再进一步合成需求的产品。工业上利用甲烷合成化学品主要通过间接转化来实现, 包括大规模制氢、合成氨[4]、合成醇[5]和费托合成等[6]。甲烷通过蒸汽重整(SRM, 式1)制备合成气是成熟的工业技术, 但仍存在催化剂结焦、中毒失活等问题[7, 8], 所以依然是催化领域的重要研究课题。甲烷蒸汽重整是强吸热过程, 也是众多甲烷间接转化过程中能耗最大的步骤。因此, 为了进一步提高过程能效, 研究者也在积极开发部分氧化(POM, 式2)、干气重整(DRM, 式3)、自热重整(ATR)和联合重整(CRM)等技术和工艺[9, 10]。其中面临的技术难题主要是催化剂的开发, 以及催化剂抗积碳、抗活性金属的烧结、抗中毒等性能的提高[11, 12]。
CH4+H2O→ CO+3H2+Δ H(1)
2CH4+O2→ 2CO+4H2+Δ H(2)
CH4+CO2→ 2CO+2H2+Δ H(3)
目前, 重整反应的催化剂主要是以贵金属催化剂和掺杂贵金属的镍基催化剂为主。甲烷间接转化过程的工艺流程比较复杂, 生产设备投资较大, 能源利用率较低, 这是工业上希望开发直接转化技术的主要原因。
甲烷直接转化是通过一步反应直接制备目的产物的过程, 主要包括甲烷氧化偶联(OCM)、甲烷选择性氧化(SOM)以及甲烷脱氢低聚(DOM), 主要产物为烯烃、炔烃和芳烃等。已经工业化的直接转化过程有甲烷高温裂解生产乙炔[13]、炭黑, 甲烷氨氧化生产氢氰酸[14]等。这些工业化的过程生产规模相对较小。而通过甲烷转化直接生产乙烯、甲醇或芳烃等大宗化工原料, 工业上急切期盼的低能耗过程却都在实验室技术开发阶段, 还需要大量的基础研究工作。
甲烷氧化偶联制乙烯技术(式4)于1982年由Keller G E等[15]首次提出, 并在全球范围内引发了研究者的广泛研究。典型的、性能较为优良的催化剂包括Na2WO4/Mn/SiO2[16]和Li/MgO[17]。但催化剂的性能特别是单程反应C2(乙烯+乙烷)产物收率和催化剂稳定性仍无法满足工业化要求。
2CH4+O2→ C2H4+2H2O+Δ H(4)
甲烷在无氧条件下的转化可以避免生成CO2和H2O等副产物, 从而提高碳原子利用率。1993年, Wang L等[18]首次报道了在Mo/HZSM-5催化剂上甲烷有效转化成苯的反应(式5), 从而在全球引发了一波针对甲烷直接脱氢低聚制备芳烃催化剂的研究。HZSM-5、MCM-22分子筛负载Mo、Re等活性金属组分的催化剂对该反应表现出良好的催化活性。
6CH4→ C6H6+9H2+Δ H(5)
甲烷选择性氧化制甲醇无论从清洁能源生产还是化学品合成考虑都是工业上梦寐以求的过程。有研究预测[19], 利用气固相催化技术, 分子氧作氧化剂, 甲烷单程转化率达到5.5%, 甲醇选择性达到80%就可以和工业上经合成气合成甲醇的过程进行竞争。然而, 文献报道的单程甲醇收率大多低于2%。所采用的催化剂活性组分多为V、Mo、Fe等的氧化物。
2CH4+O2 → 2CH3OH+Δ H(6)
经过几十年的研究与发展, 甲烷活化与催化转化的相关基础和应用研究已十分深入, 但仍存在一些关键技术难题, 致使甲烷转化效率较低, 无法达到工业化过程的要求。这些技术难题包括临氧转化过程中的选择性氧化问题、脱氢低聚过程中的热力学平衡限制、高温反应条件下催化剂失活和材料的稳定性问题。
甲烷临氧转化过程包括甲烷催化重整制合成气、甲烷氧化偶联制乙烯、甲烷选择性氧化制甲醇等。其中甲烷重整制合成气, 包括蒸汽重整、CO2干重整、部分氧化自热重整等是比较成熟的工业化技术。基于热力学原理考虑, 该技术实现工业化的主要原因之一是产品合成气CO+H2在反应体系中是相对稳定的目的产物, 并和反应物CH4+H2O处在接近化学平衡的状态, 且转化过程是热力学有利的。反观甲烷氧化偶联制乙烯和甲烷选择性氧化制甲醇, 尽管反应在热力学上有利, 但在大多数研究的反应条件下[前者(700~900) ℃, 后者(300~600)℃]反应的目的产物乙烯或甲醇不稳定, 极易和反应物中的O2发生深度氧化反应生成热力学上更加稳定的CO2或CO。这一理论事实决定了如何在反应过程中保持目的生成物乙烯或甲醇的相对稳定, 并使其及时脱离反应体系成为获得较高目的产物产率的关键。
多数文献认为, 甲烷在氧化偶联催化剂上的活化可以分为两类:一类是分子氧在催化剂表面首先被活化(如在Li/MgO上)产生亲电活性氧物种如O-或O2-, 甲烷分子与这些亲电氧物种作用发生C— H键均裂生成CH3· , CH3· 在气相发生偶联生成C2H6进而脱氢生成C2H4; 另一类是甲烷分子吸附在路易斯酸-碱对上(如在碱性La2O3或MgO表面)发生C— H键激化异裂生成甲基阳离子或在分子氧作用下生成CH3 。研究表明, 甲烷在路易斯酸-碱中心上的活化与中心的酸碱强度和局部结构有关。甲烷氧化偶联实质是甲烷脱氢偶联生成C2H6/C2H4的吸热反应与氢气氧化放热反应的耦合过程。这种反应的耦合使得甲烷脱氢生成乙烯的过程由热力学不利转化为热力学有利。然而, 由于产物或中间活性碳物种的深度氧化反应, 无法同时获得较高的甲烷转化率和产物选择性, 使产物收率受到限制。目前, 甲烷氧化偶联反应的C2收率及选择性仍无法达到工业化要求。如要获得较高的乙烯收率, 一方面需要催化剂上表面活化反应保持高度的选择性, 避免碳氢表面物种的深度脱氢和氧化; 另一方面也需要避免中间产物在气相或催化剂表面的深度氧化反应。
甲烷选择性氧化制甲醇是另一个放热且热力学有利的反应, 也是工业上梦想的大规模直接液化甲烷的催化过程。早期研究最多的是Mo-、V-氧化物催化剂[20, 21]。此类催化剂上甲烷通过与催化剂表面的MOx相互作用形成H-M(Ox)-CH3进而转化为甲醇。而近期许多研究则主要集中于模仿甲烷单加氧酶的分子筛负载Fe、Cu催化剂体系[22, 23]。为获得较高的产物收率常使用H2O2代替分子氧, 如Cui X等[24]报道的使用石墨烯限域的铁单原子催化剂上CH4低温选择性氧化反应。该类催化剂与Mo-、V-氧化物类似, 即首先在金属中心形成活性氧中间物种, 进而与甲烷分子作用使甲烷C— H键活化。目前固体催化剂上甲烷选择性氧化制甲醇所面临的最大挑战是无法同时获得较高的转化率和产物选择性。Latimer A A等[25]最近提出一个动力学模型, 认为甲烷选择氧化制甲醇连续反应过程中甲醇收率受本征动力学限制。因此, 开发温和反应条件下的氧化过程, 并对生成的甲醇予以“ 保护” 是未来的研究需要重视的[26]。
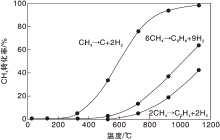
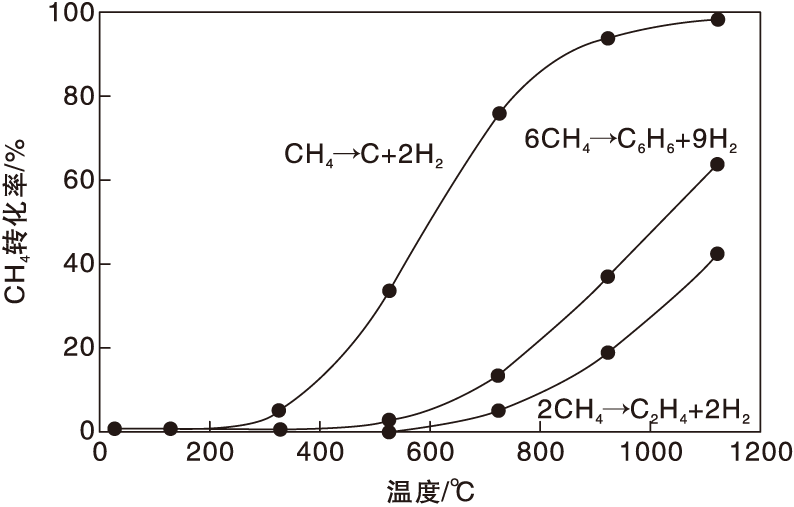
甲烷脱氢是强吸热反应, 在不同温度下甲烷转化为乙烯和苯的平衡转化率如图1所示。由于苯分子的特殊电子结构和对称性, 使得其在高温条件下比乙烯更为稳定, 在一定反应温度下甲烷转化为苯可获得更高的转化率。但即使在973K的反应温度下, 甲烷的平衡转化率也只有11.4%, 属于热力学上不利的反应。事实上, 文献研究所获得的甲烷转化率只有8%~10%, 与热力学计算结果吻合。在图1中也给出了甲烷裂解完全脱氢反应的平衡转化率。随着体系反应温度的升高, 甲烷向积碳转化的平衡转化率远远高于生成乙烯或苯的平衡转化率, 这使得反应体系中的积碳反应难于避免, 成为阻碍甲烷高效转化为烃类产物的主要难题。值得指出, 即使在高温反应条件下, 单程反应也无法获得较高的甲烷转化率, 甲烷直接芳构化过程在工业上的应用必然面临产物和反应物分离所导致的高能耗和高生产成本的问题。
在甲烷脱氢低聚反应中, 随着反应时间和反应温度的增加, 烃类热裂解易产生大量积碳, 积碳主要覆盖在催化剂的活性位上和分子筛孔道中, 导致其快速失活, 反应活性降低。这是有关甲烷脱氢低聚生产烯烃或芳烃所面临的另一方面技术难题。研究表明, 在Mo/HZSM-5催化剂上甲烷脱氢芳构化反应主要生成三种类型的表面积碳:(1)主要分布于分子筛孔道中的一种不定型或石墨型的积碳; (2)主要分布于分子筛外表面的Mo2C中的碳元素; (3)贫氢的sp型(稠环芳烃)积碳。在甲烷活化过程中, 贫氢型积碳会随着反应时间的推移而在催化剂表面逐渐累积, 直至完全覆盖活性金属和分子筛表面。针对积碳问题, 研究者使用脱铝、表面硅烷化、构建介孔结构等多种手段对催化剂进行改性研究。Ding W等[28]使用大量的外来硅源选择性地覆盖和消除沸石的外表面酸性位点, 可有效降低HZSM-5外表面酸性位点的密度, 提高单环芳烃产率, 降低催化剂失活速率。Liu H等[29]通过实验证实, HZSM-5分子筛表面经硅烷化处理后, 能够减少表面沉积的碳物种量, 从而使得催化剂反应活性提高。通过创建介孔, Wu Y等[30]将Mo浸渍到多层载体材料MFI中合成中孔/微孔沸石催化剂。结果表明, 中孔结构使反应物向孔道内活性位点的扩散更易发生, 因此在Mo/MFI催化剂上可获得更高的甲烷转化率和芳烃产率。但催化剂改性只能改善甲烷转化率和积碳问题, 无法从根本上根除积碳反应并使产物的生成突破反应热力学平衡的限制。
甲烷脱氢低聚生产烯烃或芳烃过程面临高温反应条件下产物平衡产率低以及催化剂在高温反应条件下积碳失活等技术难题。因此, 如何在较温和条件下实现较高单程反应转化率并保持催化剂一定的稳定性成为未来研究开发需要思考的问题。解决这一矛盾的对策包括:采用膜反应器将脱氢反应与氢分离过程耦合提高甲烷转化率, 以及利用氧化剂或氢捕获剂降低反应体系氢分压从而提高目的产物收率。采用膜分离反应器的有效性已被许多研究工作所证实[31, 32]。但甲烷转化率的提高却受到所采用分离膜材料氢分离性能的限制。最近, Kumar A等[33]报道在甲烷芳构化过程中添加氢吸收剂(Zr)使得甲烷在973 K下的转化率由10%提高到27%。毫无疑问, 这种分离氢的方式比采用氢分离膜更加有效, 为脱氢过程的研究提供了新思路。又例如, Kikuchi E等[34]将Pd或Pd-Ag复合膜镀在多孔氧化铝陶瓷的外表面, 突破了甲烷重整制氢的热力学平衡, 实现了氢气选择性增强的效果。研究证明在500 ℃、101.325 kPa条件下Pd膜及Pd-Ag复合膜的甲烷转化率能达到60%以上, 而理论平衡值仅为42%, 并且甲烷转化率随Pd膜厚度降低而提高。Cao Z等[35]利用钙钛矿Ba0.5Sr0.5Co0.8Fe0.2O3-δ (BSCF)膜研究甲烷芳构化反应, 发现通过氧渗透膜渗入的氧气不会破坏催化剂活性组分Mo2C。在长达1000 min的反应后, 与固定床反应器相比, 膜反应器中的甲烷转化率高出3%, 芳香族化合物选择性高出30%, 并且生成的水蒸气能有效抑制积碳, 积碳量仅占废催化剂的1.2%。Morejudo S等[36]设计了BaZr0.7Ce0.2Y0.1O3-δ (BZCY72)膜, 使氧离子迅速向反应介质传输将H2氧化为H2O, 同时操作过程中产生了少量的CO, 两者都降低了催化剂表面的结焦行为并提高了芳烃选择性。
实现甲烷在固体催化剂作用下的高效定向转化的前提条件是催化剂上的活性中心应具备适当的化学环境和空间环境, 以有利于C— H键的断裂, 或者能够产生活性中间物种进而与甲烷分子作用导致C— H键断裂。然而仅仅通过对C— H键的活化还难以达到甲烷分子定向转化为C2+烃类或甲醇等含氧有机分子的目的。碳氢键活化后所产生的CH3 、$CH_3^+$或CH2等活性中间物种必须发生相互作用生成C— C键或与第二种反应物(如分子氧或其活化后的中间物种)发生相互作用生成C— O键才可能生成目的产物。这种活性中间物种之间及其与催化剂活性中心之间的相互作用是决定甲烷优势转化方向的关键步骤。因此可以推测, 在甲烷脱氢低聚生成C2+烃类产品过程中, 形成C— C键的优势途径取决于CHx活性中间物种的相对稳定性(减少深度脱氢)和其在局部空间或催化剂表面上的浓度。因此通过构筑适当的活性中心实现C— H键的活化, 调控CHx与活性中心的相互作用并保持其适当的稳定性才有利于C— C的形成, 同时也有利于抑制深度脱氢积碳反应的发生。另一方面, 在临氧反应过程中, 无论是甲烷氧化偶联还是选择性氧化制甲醇, 都需要考虑如何避免目的产物深度氧化的问题。事实上, 甲烷临氧转化过程均可看作是甲烷脱氢反应和氢氧化反应的耦合, 而在芳构化过程中引入CO2或SO2等弱氧化剂, 也是芳构化和“ 清碳、除氢” 反应的耦合。这种体系既涉及不同反应之间能量传递过程的耦合也涉及反应物-生成物之间的功能互补。然而, 从反应耦合视角来研究甲烷脱氢转化过程的工作还远远没有深入, 值得进行广泛而深入的探讨。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|