
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TEMPO催化醇氧化反应进展
[张天琦 , 赵茂旭, 戎梅竹
, 赵茂旭, 戎梅竹* ]
, 赵茂旭, 戎梅竹]
|
|


作者简介:张天琦,1996 年生,女,在读硕士研究生,研究方向为绿色化学工艺。E-mail:tianqizhang@hotmail.com
选择性催化氧化醇类化合物为相应的醛或酮是一类重要的官能团转化反应。四甲基哌啶氧化物(TEMPO)是一种含有稳定的氮氧自由基(NO·)的有机小分子催化剂,NO·可通过自身的强选择性,在加快醛或酮转化的同时不会过氧化成为羧酸。本文阐述了TEMPO催化体系催化醇选择性氧化反应的机理,在此基础上详述了过渡金属/TEMPO、非过渡金属/TEMPO、固载化TEMPO等体系催化醇选择性氧化反应的研究情况,并探讨了TEMPO在电催化以及光催化方面的应用。指出将高活性的TEMPO催化体系改进,在发挥其高选择性、高活性优点的同时,克服其本身价格昂贵的缺点,实现催化体系的重复利用,更符合生态、绿色理念,也是未来发展的趋势。
Selective catalytic oxidation of alcohols to corresponding aldehydes or ketones is an important functional group conversion reaction.2,2,6,6-Tetramethylpiperidinooxy(TEMPO) is a small organic molecule catalyst with stable nitroxyl radicals(NO·),which can accelerate the conversion of aldehydes or ketones without peroxidation to carboxylic acids through its strong selectivity.In this paper,the mechanisms of selective oxidation of alcohols catalyzed by TEMPO catalytic system were discussed.On this basis,the research situation of selective oxidation of alcohols catalyzed by transition metal/TEMPO,non-transition metal/TEMPO,and immobilized TEMPO were detailed,and the application of TEMPO in electro-catalysis and photo-catalysis was also explored.The paper pointed out that the TEMPO catalytic system with high activity should be improved,and the TEMPO catalytic system should not only give play to its advantages of high selectivity and high activity,but also overcome its disadvantages of high price and realize the catalytic system reuseability,which is more accord with the ecological and green concept.It is also the trend of future development.
选择性催化氧化醇类化合物为相应的醛或酮是一重要的官能团转化反应。其产物醛或酮在生物、医药、香料、合成纤维等领域发挥着不可或缺的作用。四甲基哌啶氧化物(TEMPO)是一种含有稳定的氮氧自由基(NO· )的有机小分子催化剂, NO· 可通过自身的强选择性, 在加快醛或酮转化的同时不会过氧化成为羧酸。这一具有强氧化性能的活性中心被广泛应用于催化醇选择性氧化反应中。TEMPO具有价格昂贵、不易回收的缺点, 但改性后的TEMPO催化体系可实现高活性的同时易与产物分离再利用。本文简介TEMPO催化体系催化醇选择性氧化反应的机理, 在此基础上阐述了过渡金属/TEMPO、非过渡金属/TEMPO、固载化TEMPO等体系催化醇选择性氧化反应的研究情况, 并介绍TEMPO在电催化以及光催化方面的应用。
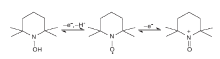
目前, 普遍认同TEMPO 选择性催化氧化醇的反应活性中心是基于其结构中氧铵盐形态的NO· 。自由基中未配对的电子在N-O键之间离域, 离域能为125 kJ· mol-1[1]。因此, TEMPO在与有机物发生强的吸热自由基夺氢反应时, NO· 可以稳定存在[2]。催化氧化反应初始, TEMPO失去一个电子成为TEMPO+, TEMPO+在反应过程中起氧化剂作用; 反应结束后, TEMPO+被还原为2, 2, 6, 6-四甲基-N-羟基哌啶(TEMPOH)(如图1所示)。TEMPOH结构中的O-H键解离能为292 kJ· mol-1[3], 低于大部分有机化合物的C-H键的解离能。因此, TEMPO在催化伯醇及小分子仲醇的过程中具有接近定量的产物选择性和高效的催化作用, 是一种具有高选择性的均相催化剂[4]。
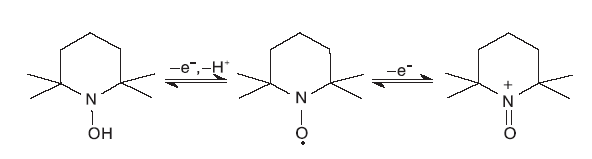 | 图1 TEMPO在催化氧化反应中的电子转移过程Figure 1 Electron transfer process of TEMPO in catalytic oxidation reaction |
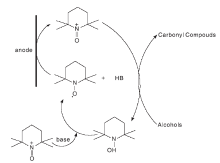
在整个反应过程中, pH值会影响TEMPO的催化性能。碱性条件下, 醇亲核进攻TEMPO+后形成的配体A是反应的关键[5]。由于醇氧离子的位阻比较大, 与TEMPO+形成配合物的难度大。因此, 在碱性条件下, TEMPO可以选择性催化氧化伯醇等位阻小的醇(图 2 所示)。
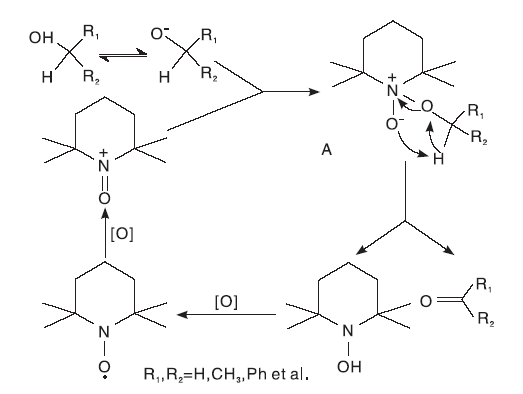 | 图2 碱性条件下TEMPO催化醇氧化机理Figure 2 Mechanism of TEMPO catalyzed alcohol oxidation under alkaline conditions |
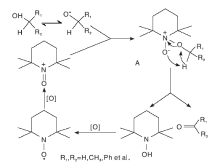
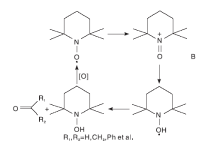
酸性条件下, TEMPO对醇的催化氧化速率比碱性条件下慢, pH< 4时, 位阻大的仲醇的反应速率高于伯醇的反应速率。Bailry W F等[6]通过计算NO· 与甲醇或异丙醇反应过程中的焓和自由能的变化, 得到了酸性条件下氢化物从醇的R-碳到NO· 中氧的双电子转移路径, 并提出了涉及中间体B的替代机理[7], 如图3所示。
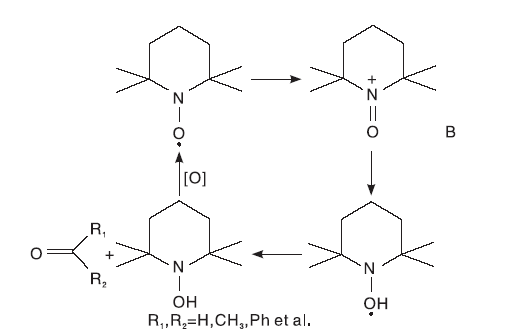 | 图3 酸性条件下TEMPO催化醇氧化机理Figure 3 Mechanism of TEMPO catalyzed alcohol oxidation under acidic conditions |
2.1.1 Ru/TEMPO催化体系
Ru配位形成的有机化合物不仅具有较高的催化活性, 同时还具有耐高温、抗氧化、耐腐蚀、强度适中等优良特性, 在醇的催化氧化反应中表现出良好的催化性能。
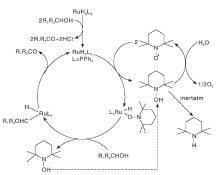
Dijkaman A等[8]报道了1%RuCl2(PPh3)3和3%TEMPO组成的催化剂, 以氯苯为溶剂, 能有效的催化氧化各种脂肪族伯醇、仲醇、苄醇和烯丙醇为醛或酮化合物, 且选择性大于99%。但该体系反应温度为100 ℃, 反应压力为1013.2 kPa, 条件比较苛刻。研究还发现, 含有S、N、O等元素的配合物对醇的反应是惰性的, 这可能是基于这些元素对Ru的配位作用造成。随后, Dijkaman A等[9]研究了RuCl2(PPh3)3/TEMPO催化体系在N2保护下对2-辛醇和苄醇的催化效果。结果表明, RuCl2(PPh3)3在反应过程中与醇反应生成了羰基化合物RuH2L3, TEMPO的作用是在重新产生Ru催化剂的同时生成TEMPOH, 如图4所示。
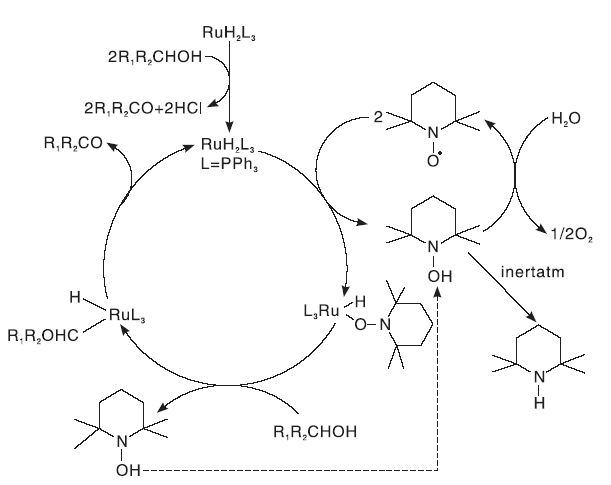 | 图4 RuCl2(PPh3)3/TEMPO催化体系的反应机理Figure 4 Reaction mechanism of RuCl2(PPh3)3/TEMPO catalytic system |
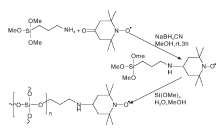
Kobayashi T等[10]将水合RuCl3负载到聚合物P1上(如图5所示), 与TEMPO在O2存在的情况下进行醇的催化氧化反应。在反应过程中, Ru的流失量最低为0.017%, 最大流失量小于0.27%。将此催化剂循环使用10次, 仍然具有很好的催化活性。
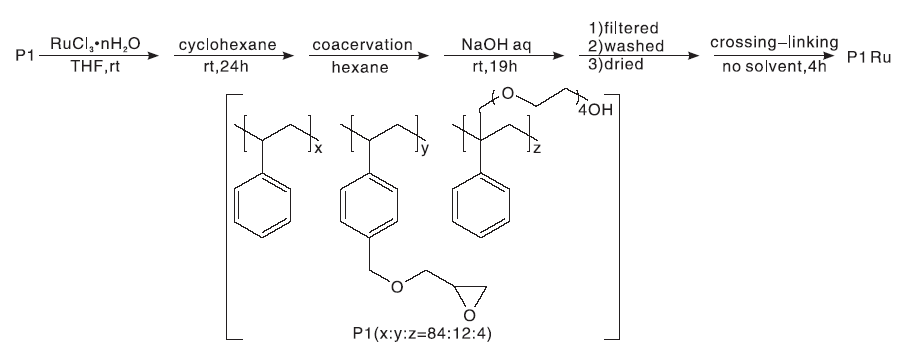 | 图5 水合RuCl3负载到聚合物P1上的制备过程Figure 5 Preparation process of hydrated RuCl3 supported on polymer P1 |
Ru催化体系在醇的催化氧化反应中已经取得了良好的研究进展, 但价格昂贵这一因素在一定程度上制约了其更为广泛的应用。因此, 开发价格低廉、催化效果较好、重复使用性高的醇氧化催化体系是实现该反应绿色、经济化的发展方向。
2.1.2 Cu/TEMPO均相催化体系
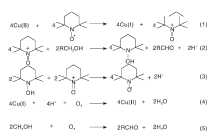
Cu价格低廉、分布较广, 可作为许多催化体系的活性中心。Semmelhack M F等[11]首次报道了CuCl/TEMPO作为醇选择性氧化反应催化体系的使用情况。结果表明, 在较温和的条件下, DMF溶剂中, 该催化体系可将伯醇选择性催化氧化成醛, 对脂肪醇的氧化无效果。该课题组认为TEMPO与Cu(Ⅱ ) 反应生成TEMPO+和Cu(Ⅰ ), 继而TEMPO+作为活性物质将醇氧化为醛, 反应后的Cu(Ⅱ ) 和TEMPO自由基可再生, 由此构成了一个完整的反应循环体系(如图6所示)。
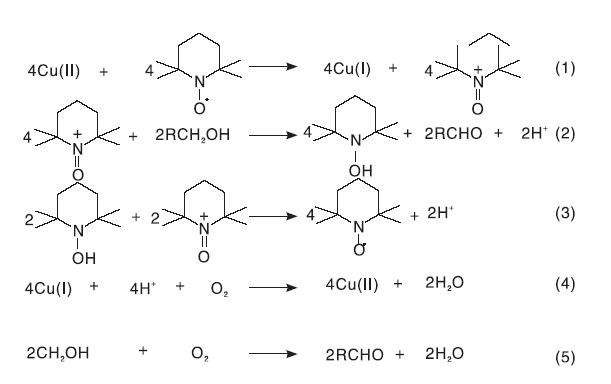 | 图6 CuCl/TEMPO催化体系的醇氧化机理[11]Figure 6 Alcohol oxidation mechanism of CuCl/TEMPO catalytic system |
Knochel B等[12]在前人研究的基础上, 对CuCl/TEMPO催化体系进行了改进, 研发出了新型的CuBr· Me2S/TEMPO催化体系, 此催化体系不但对伯醇、仲醇和烯丙基醇有较好的催化效果, 对脂肪醇也有一定的氧化效果, 但体系需要的反应温度为90 ℃。随后, 该课题组将传统的有机溶剂改变为离子液体, 同样的反应体系, 在65 ℃下, 使用摩尔分数0.005%的CuCl和摩尔分数0.005%的TEMPO便可以将苄型、烯丙型和脂族醇氧化为羰基化合物[13]。改进后的催化体系可以在相对低温的条件下实现醇的氧化, 具有节约能源、反应条件相对温和的优点。
Sheldon R A等[14]报道了乙酰-TEMPO/Cu(ClO4)2/DMAP 三组分体系。[Bmim]PF6为溶剂, 催化剂体系重复使用5次后, 活性没有明显降低。由于仲醇第二个取代基的立体位阻抑制了氧化反应过渡态的形成, 因此, 该体系对仲醇没有明显的催化效果。随后, 再该小组又研究了乙酰-TEMPO/Cu(ClO4)2/DMAP/DABCO四组分体系[15]。该体系以DMSO为溶剂, 反应结束后通过加入极性小的有机溶剂就能萃取出产物, 催化剂再生方法简单, 操作方便。
与Ru、Pd等贵金属相比较, Cu价格低廉, 实用价值较高, 将Cu催化剂不断改进成为具有底物普适性广、发展高效的催化体系其应用前景更为广泛。
2.1.3 含Fe的TEMPO催化体系
Fe元素在自然界储量丰富, 同样具有绿色清洁的优点。铁是仿生催化的一种重要金属, 研究者利用这一特性, 将铁化合物与TEMPO组成不同的催化体系, 在醇的催化氧化反应方面也取得了良好的进展。
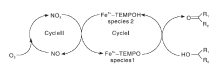
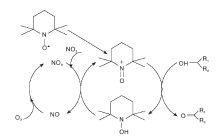
Wang N W等[16]最先研究了含Fe的FeCl3/NaNO2/TEMPO催化体系。以PhCF3为溶剂, 摩尔分数为5%的FeCl3、NaNO2和摩尔分数为2%的TEMPO为共催化剂, 空气作为氧源, 室温条件下, FeCl3/NaNO2/TEMPO催化体系可以将芳香族伯醇、含杂原子(N, S)伯醇和仲醇高选择性的氧化成为相应的醛或酮, 对脂肪族伯醇活性较低。催化体系的催化机理可以描述成两个氧化还原反应组成的循环(如图7所示)。采用化学计量的FeCl3和TEMPO催化苯甲醇的反应, 醇转化率只有35%。此结果证明:TEMPO在Fe3+的协助下经过一系列的电子转移(Cycle Ⅰ ), 引发了醇类的氧化反应, Fe3+-TEMPO(species 1)被还原生成Fe2+-TEMPO(species 2)。NaNO2产生的NO2氧化Fe2+-TEMPO为Fe3+-TEMPO的同时, 自身被还原为NO[17], Fe3+又将TEMPOH氧化生成TEMPO[18], 最终O2将NO 氧化为NO2, 完成整个催化体系的循环。
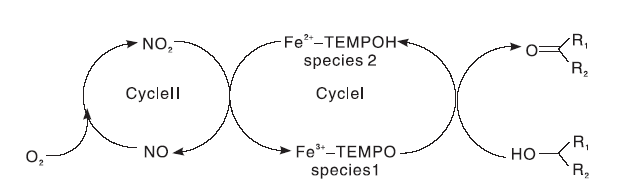 | 图7 FeCl3/NaNO2/TEMPO体系催化醇氧化机理Figure 7 Mechanism of FeCl3/NaNO2/TEMPO catalyzed alcohol oxidation |
随后, Wang N W[19]采用更经济的Fe(NO3)3/4-OH-TEMPO催化体系, 常温常压下, 以乙腈为溶剂, 可以高效、高选择性地催化醇氧化反应, 并具有更广的底物适用范围。课题组认为该体系催化醇氧化的机理与FeCl3/NaNO2/TEMPO催化体系机理相似, 也可以用氧化还原反应构成的两个循环来解释。
2.1.4 含双金属盐的TEMPO催化体系
Cecchetto A等[20]发现将Mn和Cu 两种金属结合在一起催化氧化效果更好。该课题组研究了双金属催化体系Mn(NO3)2/Cu(NO3)2/TEMPO在乙酸溶剂中对仲醇的催化效果。结果表明, TEMPO在酸性条件发生歧化反应得到氮羰基阳离子, 但该催化体系不溶于非酸性溶剂。
在此研究的基础上, Guo Yanchun等[21]采用共沉淀法制备了Cu-Mn复合氧化物作为反应体系的催化剂。实验结果表明, Cu-Mn复合氧化物的催化性能比单一分的CuO和 MnO的效果更好, 反应条件更温和, 回收方法更简便。
金属参与的催化体系在使用后会产生一定的含金属废液, 因此寻找不产生有害废物的无过渡金属参与的催化体系更符合绿色化学的理念。
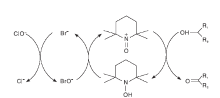
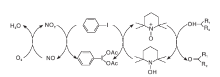
Anelli P L等[22]首先研究探讨了非过渡金属TEMPO/NaClO催化体系, 由于体系中含有部分的NaHCO3, 因此, 整个反应在弱碱性条件下进行, 在短时间内将醇氧化为相应的醛或酮。在整个催化循环过程中, NaClO将TEMPO氧化为TEMPO+, 随后TEMPO+将醇氧化为相应的醛或酮, TEMPO+则被还原为TEMPOH; 生成的TEMPOH重新被NaClO氧化为TEMPO+; 基于ClO-可以将Br-氧化为氧化性更强的BrO-。因此, 反应过程中加入了KBr以提高反应速率(如图8所示)。
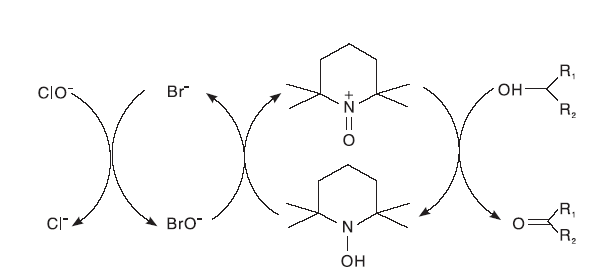 | 图8 TEMPO/NaClO体系催化醇氧化机理Figure 8 Mechanism of TEMPO/NaClO catalyzed alcohol oxidation |
基于此, 以TEMPO/NaClO催化体系为基础的衍生体系不断被研发出来。Herrerial C I等[23]研究了TEMPO/Br2/PhIO2催化体系, 利用NO来活化分子氧, 加入的乙酰碘苯实现了NO2和TEMPO之间的电子转移(见图9)。该催化体系可高效、定量的将各种芳香醇氧化为相应的醛和酮, 含N、S杂原子的底物不影响反应的进行, 伯醇的氧化产物中会有酸和酯生成。
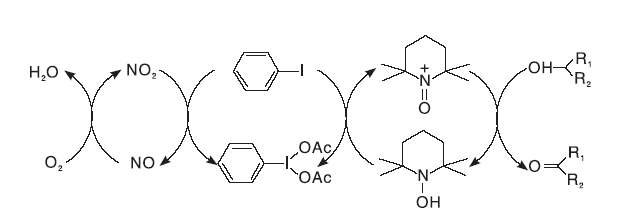 | 图9 TEMPO/Br2/PhIO2体系催化机理Figure 9 Catalytic mechanism of TEMPO/Br2/PhIO2 system |
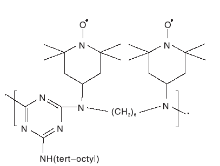
Hu X等[24]开发了TBN/TEMPO双组份催化体系。其中, TBN不稳定, 受热后很容易释放出NO或NO2, 与氧气作用后将TEMPO氧化为TEMPO+, TEMPO+迅速将醇氧化为醛或酮, 自身被还原为TEMPOH, TEMPOH又被NO2重新氧化为TEMPO+, 实现了整个催化体系的循环(如图10所示)。
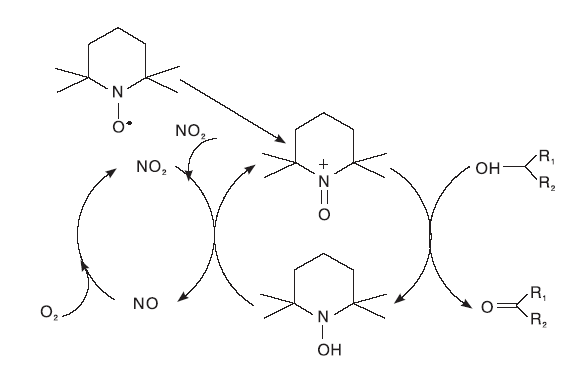 | 图10 TBN/TEMPO体系催化机理Figure 10 Catalytic mechanism of TBN/TEMPO system |
漆酶是一种多铜中心的生物仿生酶[25], 可催化氧化降解木质纤维素, 工业上常用来纸浆漂白。Arends I W C E等[26]尝试将漆酶与TEMPO催化体系用于醇的催化氧化反应, 室温、常压和pH=5条件下反应可进行。但TEMPO用量较大(20%~30%), 反应速率慢, 存在反应成本高、能耗大的问题, 工业生产中受到了一定限制。
作为均相催化剂, 价格较高的TEMPO在使用过程中还存在不易被回收的问题。因此, 将TEMPO固载后多相化, 即可以解决其价格较高的缺点又可以实现催化剂的回收再利用。常用的固载 TEMPO 的载体包括硅胶、分子筛和聚合物等。
2.3.1 硅胶固载 TEMPO 催化体系
硅胶具有多孔性、吸附能力高、稳定性好等特点。由于硅胶表面裸露羟基, 可用于固载多种催化活性中心[27]。
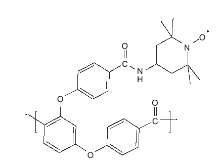
文献[28, 29]制备了SiO2-NH-TEMPO催化剂(结构如图11所示)。课题组分别以苯甲醇、4-硝基苯甲醇、2-溴苯甲醇、2-壬醇等为底物, 分别采用SiO2-NH-TEMPO和TEMPO 为催化剂进行了对比试验, 1 h后, 两种催化剂皆能得到选择性氧化产物, 固载后的SiO2-NH-TEMPO与均相TEMPO催化效果相似, 活性没有降低。
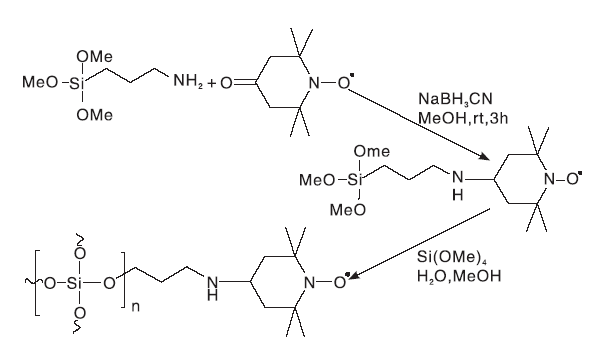 | 图11 SiO2-NH-TEMPO催化剂Figure 11 SiO2-NH-TEMPO catalyst |
2.3.2 介孔分子筛固载TEMPO催化体系
介孔分子筛具有比表面积大, 孔径分布均一, 孔径可调变等良好的结构性能。由于介孔分子筛的孔内壁上存在一定数量表面羟基缺陷, 使得某些物质可以通过与硅羟基发生反应而键合于孔内表面, 是一优异的载体材料。
Brunel D等[30]制备了MCM-41-ether-TEMPO和MCM-41-amide-TEMPO(结构如图12所示)两种MCM-41负载TEMPO的催化剂。研究结果显示, 两种催化剂均可以催化氧化α -甲葡萄糖苷成1-O-甲基葡萄糖醛, 催化效果没有差异, 且选择性大于95%。此外, 以O2为氧源, MCM-41-ether-TEMPO/CuCl催化体系与均相TEMPO/CuCl催化体系具有相似的选择性, 都大于99%, 但转化率较低, 仅为35%。
 | 图12 MCM-41-ether-TEMPO(左)和MCM-41-amide-TEMPO催化剂(右)Figure 12 MCM-41-ether-TEMPO(left) and MCM-41-amide-TEMPO catalyst(right) |
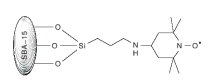
Babak K等[31]制备了SBA-15-TEMPO(结构如图13)。课题组探讨了SBA-15-TEMPO/NaNO2/nBu4NBr催化体系以O2作为氧化剂对苯甲醇的催化性能。研究发现, 99.8%的苯甲醇被选择性氧化为苯甲醛, 转化率为100%。进一步研究发现, SBA-15-TEMPO 能够在无过渡金属存在的条件下催化伯醇和仲醇, 连续循环使用14次后, TEMPO活性中心没有流失。
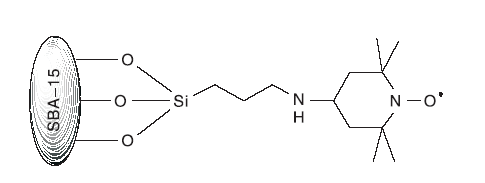 | 图13 SBA-15-TEMPO催化剂Figure 13 SBA-15-TEMPO catalyst |
2.3.3 聚合物固载TEMPO催化体系
常见的聚合物载体有聚乙二醇(PEG)、聚氨基甲酸酯(PU)、聚己内酯多元醇(PCL)等。PEG是一种可溶性聚合物载体, 具有无毒、溶解性高、易功能化、价格低廉和原材料易得等优点。Pozzi G M 等[32]和Benaglia M等[33]尝试将TEMPO负载到PEG上(如图14所示)。结果表明, 在PEG端嫁接TEMPO得到的多相催化剂具有良好的催化性能, PEG 的分子量及嫁接TEMPO的位置等因素对催化剂活性和回收率有直接影响[34], 添加适量乙醚可以使催化剂沉淀分离, 催化剂回收率高于95%。
 | 图14 TEMPO-PEG催化剂Figure 14 TEMPO-PEG catalyst |
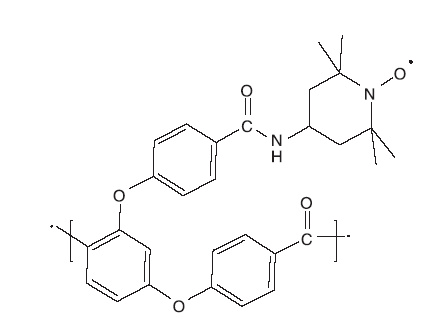
Dijksman A等[35]将TEMPO负载于可溶性低聚物光稳定剂Chimassorb 944分子上, 制成低聚物负载型TEMPO催化剂PIPO(如图15)。 PIPO/CH2Cl2/NaClO/KBr催化体系比 TEMPO的催化活性更高, 而且后期分离操作简单, 通过过滤即可回收。
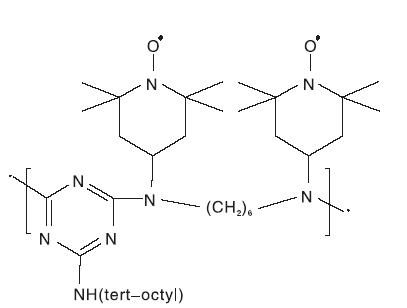 | 图15 TEMPO-PIPO催化剂Figure 15 TEMPO-PIPO catalyst |
Subhani M A等[36]以聚氨酯为载体, 将带有异氰酸基团的聚合物与4-OH-TEMPO 结合作为活性中心进行了负载, 制备了PU-TEMPO和PS-TEMPO催化剂。该催化剂可以应用于部分醇到醛的氧化, 在水/二氯甲烷/NaOCl作为助氧化剂体系中或者单相的反应体系中具有极高选择性。
通过化学键将NO· 连接到具有其他活性中心的有机配体上, 可以得到同时具有NO· 与其他活性中心的双官能团配合物。双官能团催化剂同时具有NO· 和其他活性的催化效果, 利用这种新型的双官能团的配合物催化醇选择性氧化反应是一种新的催化方法。
Lu Z等[37, 38]先后合成了三种含有联吡啶和NO· 活性中心的双官能团三嗪衍生物。在t-BuOK存在条件下, 以CH3CN和H2O为混合溶剂, 配合物与CuBr2共同作用可以有效催化苄醇的有氧氧化反应。研究结果表明, 化合物a~c(图16)均可以与铜盐原位生成络合物, 可更加高效的催化醇氧化反应。
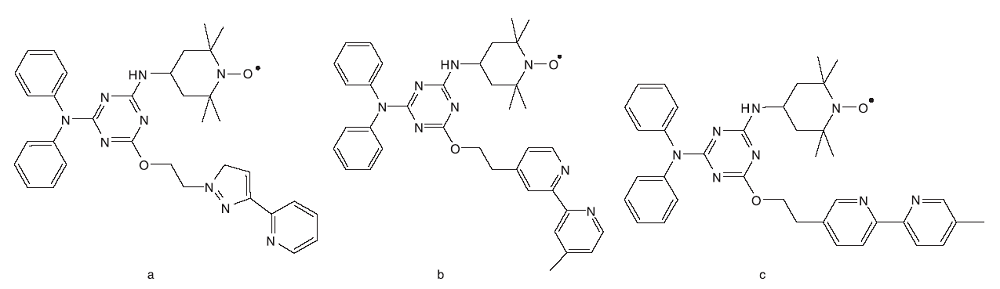 | 图16 TEMPO/N-配体双官能团化合物结构图Figure 16 TEMPO/N-ligand bifunctional compound structure diagram |
2.4.1 电催化醇氧化
催化醇氧化过程中, 需加入一定量的氧化剂, 有些氧化剂的加入会给环境带来一定程度的污染。使用电极代替传统的氧化剂, 利用电化学方法进行氧化还原反应是另一种符合绿色化学的方式。单室电解池和双室电解池都可以进行醇的催化氧化反应。在单室电解池中, 以 MeCN 和水的混合溶液作为溶剂[39]或以DCM 和水的两相体系作为溶剂[40]都可以进行电解; 在双室电解池中, 以MeCN 或者 DCM作为溶剂, 以催化化学计量的TEMPO, 可以通过控制电位或者控制电流的方式进行醇的催化氧化。在使用TEMPO进行醇的催化氧化过程中, 通过阳极氧化的方法得到TEMPO+, 反应机理如图17[41]。
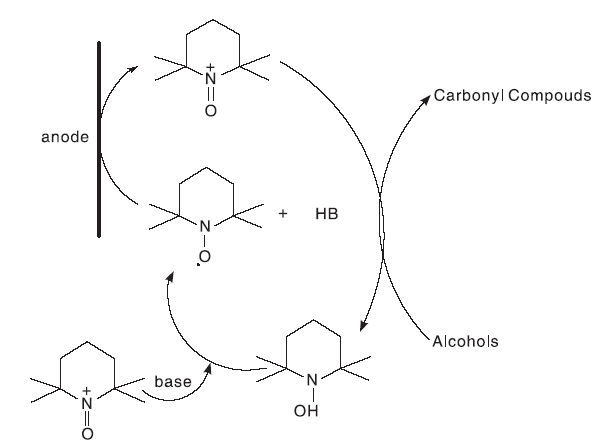 | 图17 电化学方法催化醇氧化机理Figure 17 Catalytic alcohol oxidation mechanism for electrochemical method |
离子液体是一种清洁型溶剂, 具有优异的传导性和稳定的电化学性质。Barhdadi R等[42]通过循环伏安法, 证明TEMPO在离子液体中发生了可逆的氧化还原反应, 能将醇氧化为醛的氮羰基阳离子。在很多研究中都达到接近100%的感应电流和选择性。但随着电解时间的延长, 得到烯醇式产物的同时会使催化剂失活。
2.4.2光催化醇氧化
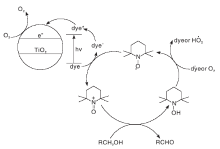
光催化醇氧化是在光照条件下, 以氧气或空气作为氧化剂, 以具有光催化效果的材料为催化剂, 催化醇氧化为相应的醛和酮[43]。Zhang M等[44]报道了经过染料敏化的TiO2在可见光照射、常压O2、无需任何溶剂条件下, 与TEMPO形成催化氧化体系, 具有节省能源、选择性高的优点, 选择性可达到100%, 反应机理如图18所示。
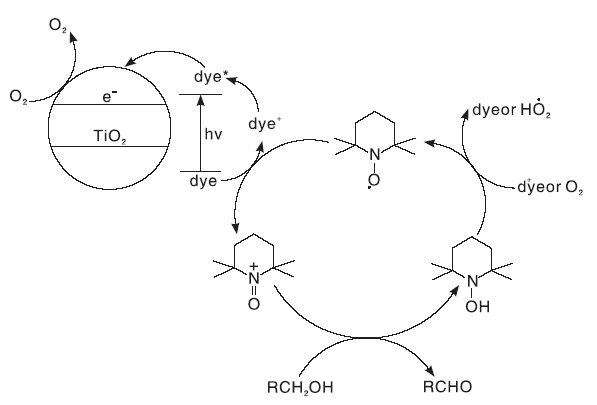 | 图18 光催化醇氧化反应机理Figure 18 Mechanism of photocatalytic alcohol oxidation reaction |
Zhao J C等[45]构建了TiO2/茜红素/TEMPO三组份催化体系, 利用TiO2将茜红素氧化得到阳离子自由基氧化性活性物种, 然后与4-羟基哌啶醇氧自由基反应, 得到具有温和氧化性的TEMPO+, 最终以O2为清洁氧化剂, 实现各类醇到醛的高选择性、高转化率氧化。
含有氮氧自由基的TEMPO与其它化合物组成的均相催化剂体系显示了优异的催化性能, 但是高昂的价格及后期分离问题限制了其进一步的工业应用。因此, 将活性组分氮氧自由基固载, 既可以解决其价格较高的缺点又可以实现催化剂的回收再利用。TEMPO的多相化更符合绿色可持续发展理念, 这也是未来发展的趋势。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|