
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
甲烷选择性氧化制甲醛/甲醇多相催化剂研究进展
[王莹1 , 孔莲1, *  , 赵震
, 赵震1, 2, * , 范晓强1 , 解则安1 , 肖霞1 ]
, 赵震, 范晓强|
|


作者简介:王 莹,1994年生,女,辽宁省朝阳市人,在读硕士研究生。
甲烷是天然气、页岩气等化石能源的主要成分,储量十分丰富。由于甲烷分子结构高度稳定,其高效活化与选择性转化具有很大的挑战性,被认为是催化反应领域的“圣杯”,因此,如何在温和条件下实现甲烷选择性氧化为高附加值的含氧化合物(如甲醇、甲醛)成为研究热点。近几十年来,研究者在甲烷选择性氧化催化剂的设计与制备方面开展了大量卓有成效的工作,但仍不能满足工业化生产要求。围绕这一反应,主要介绍甲烷通过气固相催化氧化、液固相催化氧化反应制备甲醛和甲醇的研究进展,并对这些催化体系的作用机理、活性物种类型和活性位结构进行科学认识。期望通过对甲烷选择性氧化催化剂的深入探究,最终指导高活性、高选择性催化剂的设计与开发。
Methane is the main component of fossil fuels such as natural gas and shale gas,and its reserves are plentiful.The efficient activation and selective transformation of methane molecule is a great challenge due to its highly stable structure,which is regarded as the “holy grail” in the field of the catalytic reactions.Therefore,how to realize the selective oxidation of methane to high value-added oxygenated chemicals(such as methanol and formaldehyde) under mild conditions has become a hot research topic in recent years.In decades,a lot of effective work in the design and preparation of catalysts for the selective oxidation of methane have been done.But there is still a huge gap from the requirements of industrialization.In the review,we mainly summarizes recent advances in the synthesis of formaldehyde and methanol by the gas-solid and liquid-solid catalytic oxidation of methane.And a scientific understanding of the mechanism of these catalytic systems,the types of active species and the structure of active sites has been also investigated.It is expected to provide the guidance for the design and development of high activity and high selectivity catalysts through the in-depth exploration of methane selective oxidation catalysts.
煤、石油、天然气是现代社会重要的燃料和化工原料。煤炭资源储量丰富, 分布范围广, 但是煤炭的过度开采与利用过程会产生大量的废弃物, 从而污染环境, 造成生态环境的进一步恶化[1]。石油作为全球最重要的能源之一, 对世界经济发展和政治稳定都具有重要影响。近年来, 由于石油资源地区分布不均衡、价格持续猛涨、资源日益紧缺等原因使得人们越来越关注储量丰富、洁净环保的优质能源。天然气、页岩气和可燃冰等是绿色、清洁的新碳基能源, 具有对环境友好、易燃烧、燃烧效率高等优点, 其开发与利用可在一定程度上缓解对石油等传统化石能源的依赖, 还具有重要的经济意义[2, 3, 4, 5]。
作为天然气、页岩气等资源主要组成部分的甲烷分子, 其C-H键的键能较高(439.3 kJ· mol-1)、结构稳定, 这使得甲烷在常规条件下很难活化[6, 7, 8]。因此, 甲烷的C-H键需要在相对苛刻的反应条件下才能被活化, 比如高温高压、强酸/碱; 或者采用非常规手段如等离子[9]、光激发[10]等。目前, 工业上甲烷的活化与利用主要采用间接转化法, 但此方法流程复杂, 能耗大, 生产成本高[11, 12, 13, 14], 因此, 寻求经济、高效的甲烷转化途径势在必行。
甲烷选择性氧化可以一步得到具有高附加值的含氧化合物, 如甲醇、甲醛, 此路径比间接合成路线更为节能, 设备更简单; 同时能使天然气、页岩气等资源得到合理、高效的利用, 因此甲烷的直接转化利用具有较大的市场前景和应用价值[15, 16, 17, 18, 19, 20, 21]。但已报道的大多数催化剂仍很难打破“ scaling relationship” 限制, 催化剂活性与选择性仍需进一步提高。
甲烷选择性氧化制含氧化合物的反应体系主要包括:均相催化反应体系[22, 23, 24, 25, 26, 27, 28, 29, 30]、气-固相反应体系[31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41]、液-固相反应体系[42, 43, 44, 45, 46, 47, 48, 49, 50]、生物法[51, 52, 53, 54]以及光催化反应法[55, 56, 57, 58]。一般来说, 多相催化剂比均相催化剂更适用于下游的工业应用, 因为催化剂更易回收再利用。本文详细阐述近年来甲烷分子选择性氧化制含氧化合物在气-固相反应体系和液-固相反应体系的研究进展, 分析这两类反应体系中催化活性位的结构和催化反应机理, 对各类催化体系的优缺点进行总结, 以期望为甲烷选择性氧化反应的深入研究提供理论指导, 并对甲烷选择性氧化反应的发展前景进行展望。
多相催化反应体系一直是甲烷催化氧化反应的研究热点, 这是由于均相催化剂价格昂贵, 难于回收利用, 还会造成严重的环境污染。多相催化反应体系包括气固相催化氧化反应和液固相催化氧化反应。新催化体系的探索、催化反应机理、催化剂构效关系的研究是气固相催化氧化反应的主要研究内容。这类催化反应体系中, 多以负载型金属氧化物催化剂的研究为主, 常用O2或N2O作为反应的氧化剂[59, 60]。但在不同氧化气氛中, 甲烷选择性氧化产物会有差别, 如O2(强氧化剂)会使一部分CH3OH氧化为HCHO而形成二者的混合产物, 而N2O(弱氧化剂)气氛中更倾向于生成HCHO。由于产物不同, 所使用的催化剂体系也会有所差别, 因此根据产物类型将气固相催化反应体系分为两大类, 即甲烷选择性氧化制甲醇和甲烷选择性氧化制甲醛。
甲醇不仅是应用广泛的基本化工原料, 还是性能优良的能源和汽车燃料。从热力学角度看, 甲烷选择性氧化制甲醇反应可以在室温下自发进行(CH4+0.5O2→ CH3OH, Δ G298K=-111 kJ· mol-1), 但是产物甲醇(C-H键能为393 kJ· mol-1)比反应物甲烷更活泼, 易深度氧化为
1.1.1 Mo基催化剂
早在1965年, Atroshchenko V I等[63]就发现MoO3在高温高压条件下可以使甲烷部分氧化为甲醇, 而且甲醇在催化剂上的连续氧化非常困难。随后Dowden D A等[64]继续对Mo基多组分氧化物催化剂进行研究, 发现将MoO3负载到载体上可以提高其甲烷选择性氧化性能; 且反应压力的增加, 有利于操作温度的降低。以25%Al2O3/75%SiO2为载体负载MoO3催化剂, 在反应温度为(403~773) K, 压力0.5 MPa时, 甲醇产率为869 g· (kg-cat· h)-1。Zhang X等[65]对比研究不同载体(ZrO2和La-Co-O)负载MoO3催化剂上甲烷选择性氧化制甲醇时发现, 以ZrO2为催化剂载体仅能产生痕量甲醇, 而La-Co-O负载质量分数7%MoO3催化剂上甲烷转化率为11.2%时, 甲醇选择性可达到60%。表征结果表明, 适中的还原性和O-/O2-有利于甲醇选择性的提高, 而[MoO4]2-物种则是甲烷选择性氧化为甲醛的活性物种。
除了载体效应外, 反应气氛中存在水蒸汽也会影响催化剂的反应性能。Liu R S等[66]研究了MoO3/SiO2催化剂上甲烷选择性氧化制甲醇反应, 结果表明, 在原料气中加入水蒸汽可以提高甲醇选择性, 但水蒸汽的引入会使活性Mo物种转变为氢氧化物, 从而限制了这类催化剂的工业应用。Ayako K与Sugino T等[67, 68]的研究成果也证明MoO3/SiO2催化剂在有过量水蒸汽存在时, 可以提高甲烷选择性氧化反应的活性。这是由于反应过程中SiO2表面形成了钼酸盐(SMA∶ H4SiMo12O40), 这是催化反应的活性物种, 它能抑制CH3OH等产物深度氧化为COx。此外, 还进一步探究了制备方法对MoO3/SiO2催化剂催化性能的影响, 结果表明, 与浸渍法相比, 溶胶-凝胶法制备的催化剂具有更好的反应性能[67]。当反应温度为873 K时, 甲烷转化率和甲醇选择性分别为8.2%和11%。
由于Mo基催化剂通常在较高温度或有水蒸汽存在条件下才能产生甲醇, 因此近年来有关这类催化剂的报道较少。
1.1.2 V基催化剂
过渡金属钒具有多变的价态和强的氧化-还原能力, 能够较好地活化氧分子形成活性氧物种, 从而实现对甲烷分子的活化与转化。助剂、制备方法和钒负载量是影响V基催化剂甲烷选择性氧化性能的重要参数, 前者决定了反应的工艺条件, 后两者决定了活性钒氧化物的结构和性质。
添加剂能降低反应的起始温度, 并提高甲醇的选择性[69]。Barbero J A等[70]研究发现, 在原料气中添加少量的NO改变了反应物中CH3· -CH3O2· 自由基的比例, 促进了甲烷的活化, 从而提高产物选择性。当NO浓度为1%、反应温度为923 K、CH4与O2物质的量比为1.8时, 甲烷转化率约为40%, 甲醇和甲醛总选择性高达40%。但是甲烷选择性氧化反应由于O2存在, 不可避免地会在产物中生成H2O, NO和H2O共存时会腐蚀反应设备; 而且仍然需要在较高温度才能获得理想的催化性能。
为了明确SiO2负载钒催化剂中活性位的结构, Wang C B等[71]首次报道了以两步溶胶-凝胶法制备具有大比表面积的V2O5-SiO2干凝胶上甲烷氧化反应, 深入研究了催化活性位的结构、性质与催化剂性能之间的构效关系。结果表明, 随着钒负载量的增加, 表面可触及的四面体V5+物种逐渐转变为聚合的V5+物种, 后者不能使甲烷转化为含氧化合物。当钒负载质量分数量为1%时, 甲烷转化率为0.77%, 而甲醇转换频率为4.0× 10-2s-1。
从这些研究结果不难发现, 钒基催化剂和钼基催化剂相似, 在高温、高压或者添加助剂时才能获得高甲烷转化率和高甲醇选择性。因此, 研究者们通过寻求新的催化剂体系来实现温和条件下甲烷的高效活化与转化。
1.1.3 其他催化剂
自然界中甲烷单加氧酶催化剂(MMO)可以在温和条件下实现甲烷的活化与转化, 在较高甲烷转化率时甲醇选择性约100%[72, 73, 74, 75, 76], 这为研究者开发高效的低温甲烷选择性氧化催化剂提供了新的思路。制备金属分子筛催化剂模拟MMO的结构, 形成活性的双核铁或双核铜中心, 以期望实现温和条件下甲烷的高效转化。
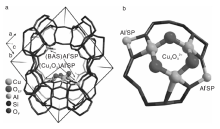
在这一类催化剂中, 以Cu为活性组分的Cu-分子筛催化剂可以在O2气氛中将甲烷直接氧化为甲醇, 但是Cu-沸石分子筛催化剂中活性物种的归属仍然存在争议。21世纪初期, 研究者首先将Cu-ZSM-5催化剂在723 K的O2气氛中预处理一段时间直接与甲烷气体反应, 实现了甲烷与O2无直接接触产生CH3OH[77]。后续研究确认, Cu-ZSM-5催化剂中只有弯曲的双核[Cu(μ -O)-Cu]2+物种是催化活性位, 而不是之前认为的Cu2O2结构[78]。Grundner S等[79]研究发现, 以丝光沸石(MOR)为载体可以改变Cu-分子筛催化剂中活性物种的结构, 双核[Cu2O]2+物种转变为([Cu3(μ -O)3]2+)三聚铜氧团簇, 其结构如图1所示。
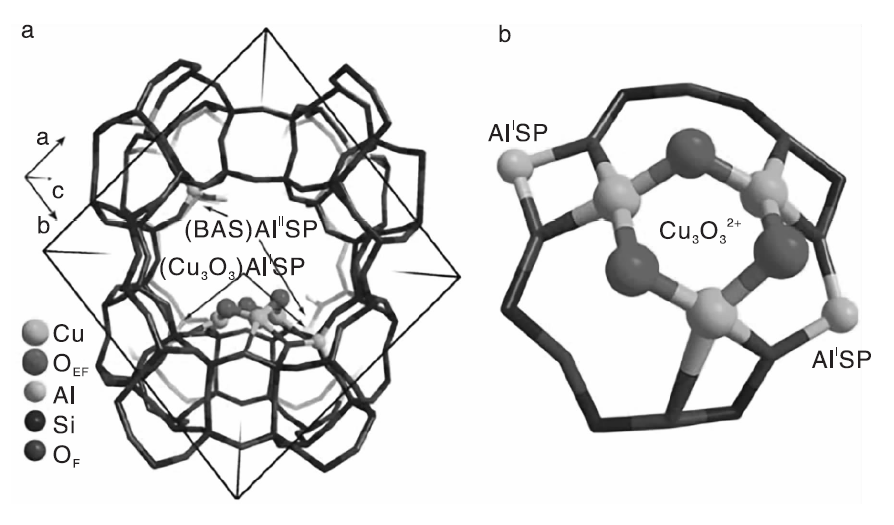 | 图1 DFT计算MOR沸石中[Cu3(μ -O)3]2+三聚铜氧团簇的位置(a)和结构(b)[79]Figure 1 Location (a) and structure (b) of [Cu3(μ -O)3]2+cluster in mordenite calculated by DFT[79] |
三聚铜氧团簇结构非常稳定, 且在甲烷转化为甲醇反应中表现出较高的催化活性, 甲醇总收率可高达160 μ mol· g-1, 高于相同条件下Cu-ZSM-5催化剂上甲醇收率[78]。此体系虽然获得较高的甲醇收率, 但不可避免存在甲烷与O2直接接触发生爆炸的危险。
Kim Y等[80]采用弱氧化剂N2O代替O2时发现, 在N2O气氛中活化时, 94%的非活性Cu-O物种转变为活性三聚铜氧团簇Cu3(μ -O)3]2+, 873 K时甲醇产率是O2气氛下(723 K活化)甲醇产率的1.5倍。但从生产成本看, 相比O2, N2O来源不易, 且价格昂贵, 因此限制了其工业应用。助剂也会影响催化剂的反应性能, Tomkins P等[81]研究铂钯掺杂Cu-MOR催化剂上甲烷等温直接转化甲醇的反应性能时发现, 贵金属Pt和Pd前驱体和铜物种聚集形成双金属铜氧团簇, 从而有助于铜氧化物的还原, 这使得活性较低的位点能够参与反应。与传统Cu-MOR催化剂相比, PtCu-和PdCu-MOR催化剂在低温活化比高温活化的催化性能更高。虽然经过长时间研究探索, Cu-分子筛催化剂的甲烷选择性氧化性能得到较大程度提高, 但对其反应活性位结构的指认仍是研究重点。近年来研究表明, Cu-SiO2催化剂也可以在O2存在条件下逐步将甲烷转变为甲醇, 而不采用沸石分子筛作为催化剂载体[82]。虽然Cu-SiO2催化剂在1 073 K才能获得与Cu-MOR和Cu-ZSM-5催化剂在673 K时相同的甲醇产率, 但这项研究结果打破了以往认为只有在结晶沸石分子筛载体上才能形成活性铜氧物种的观点。因此, 只要条件合适, 许多其他载体也可能用于负载过渡金属Cu, 从而催化甲烷高效转化为甲醇。
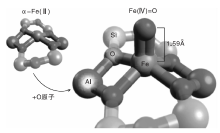
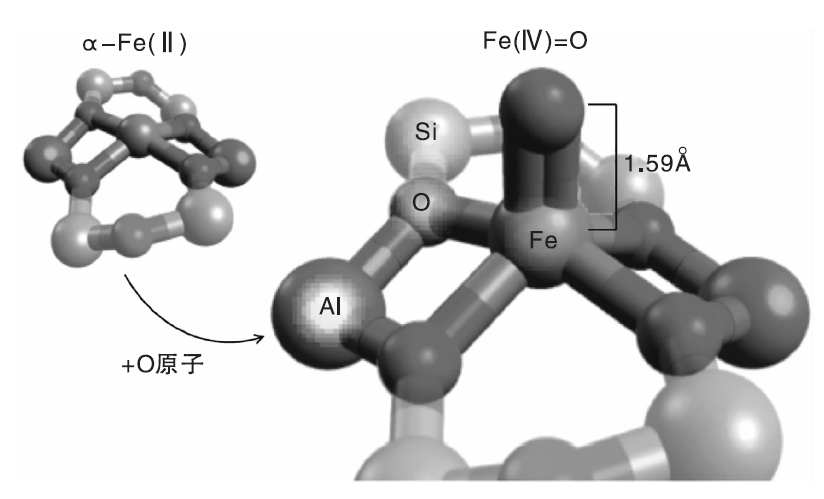
以过渡金属Fe为活性组分的类MMO催化剂则需要在N2O气氛中才能高效地活化甲烷分子, N2O首先在催化剂的α -Fe(Ⅱ )位点上分解产生所谓的α -O物种, 然后该物种与甲烷进一步反应。但是Fe-分子筛催化剂上活性位的性质以及决定其异常反应性能的因素多年来一直没有定论[83]。Snyder B E R等[84]利用变温变场圆二色谱(VTVH-MCD)表征和理论计算对Fe(Ⅱ )-BEA催化剂中的铁物种进行研究, 发现反应活性位是单核、高自旋的平面四边形Fe(II)物种[α -Fe(Ⅱ )], 反应过程中该物种得到N2O中的氧原子形成单核、高自旋的反应中间体— — [FeO]2+物种[Fe(Ⅳ )=O], 其结构如图2所示。该活性物种具有高活性和选择性的原因在于沸石分子筛晶格对其具有配位约束作用。Parfenov M V等[85]研究表明, Fe-ZSM-5催化剂上甲烷氧化制甲醇反应也可以在“ 准催化” 模式下进行, 通过比较“ 准催化” 和催化条件下的反应结果可以获得关于反应机理的信息; 还发现在体系中引入水蒸汽可以提高CH3OH的选择性。反应温度为573 K时, 未添加水蒸汽的体系中甲醇选择性仅1.9%, 而添加30%水蒸汽, 甲醇选择性提高到42%。从上述研究中获得的前期知识可以启发研究者设计新颖、高效的模拟MMO中催化活性位点的无机催化剂。但是到目前为止, 金属分子筛催化剂的制备方法以及分子筛的结构对活性物种结构和性质的影响仍不明确, 因此需要更加深入地研究催化活性位的微观结构和局部环境对催化性能的影响, 从而为设计和开发高效甲烷选择性氧化催化剂打下坚实的基础。
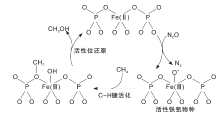
此外, 体相或负载型FePO4催化剂在N2O气氛中也可以有效地活化甲烷分子, 这些催化剂在反应温度673 K时, 产物CH3OH和HCHO选择性可高达90%[86, 87]。研究表明, 表面还原的Fe(Ⅱ )可以活化N2O分子产生活性铁氧物种Fe(Ⅲ )-O-, 从而活化甲烷(如图3)。对比研究FeOx/SBA-15催化剂上甲烷选择性氧化性能发现, Fe位点周围的磷酸基是CH3OH形成的必要条件[88, 89, 90]。在以
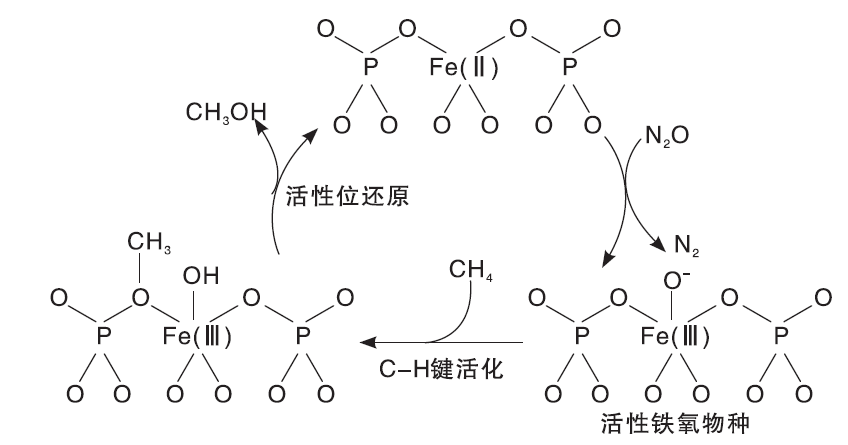 | 图3 N2O气氛中, 体相或负载型FePO4催化剂上甲烷氧化制甲醇的反应机理[89]Figure 3 Tentative reaction mechanism for the selective oxidation of methane to methanol with N2O as oxidant over bulk FePO4 or supported FePO4 catalysts[89] |
尽管目前有较多的关于甲烷制甲醇气固相催化反应的研究, 且具有类MMO结构的过渡金属分子筛催化剂也实现了温和条件下甲烷的高效转化, 但仍存在甲烷转化率和甲醇收率远不及工业化要求的问题。因此, 要实现甲烷气固催化氧化反应的工业化, 关键在于设计并研制出高效的催化剂, 提高甲烷转化率的同时增加甲醇选择性。
甲醛是一种应用广泛的C1平台化合物, 工业上主要由甲醇氧化[反应温度(873~973) K]或天然气直接氧化[反应温度(873~953) K]制得。目前, 甲烷选择性氧化制甲醛反应比较困难, 甲醛产率基本< 4%, 原因在于高温、氧化气氛中甲醛极易被进一步氧化为COx, 使得高活性与高选择性难以兼得, 因此甲烷选择性氧化制甲醛仍是催化领域乃至化学领域的“ 圣杯” 反应[91]。通过对各种各样的催化剂上甲烷选择性氧化制甲醛反应性能及反应机理的研究, 发现活性位和载体结构调控、制备方法改进都可以显著改善催化性能, 但是高的甲醛产率仍然难以获得。所报道的催化剂中, 过渡金属V、Mo、Fe氧化物催化剂具有较好的催化性能。
1.2.1 Mo基催化剂
过渡金属Mo具有可变的化学价态, 在反应过程中可以提供晶格氧形成氧空穴或者还原形成吸附氧物种, 因此形成高、低化合价之间的氧化还原循环, 遵循Mars-Van Krevelen氧化-还原机理。其中, Mo含量和Mo-O物种结构的变化对催化剂的性能影响较大。一般来说, 甲烷转化率随Mo含量的增加而增大, 而甲醛选择性则存在一个极值范围。Banares M A等[21]研究了Mo负载量对MoO3/SiO2催化剂上甲烷氧化制甲醛性能的影响, 结果表明, 随着Mo原子密度增加, 甲醛选择性呈“ 火山型” 曲线变化规律。当Mo原子密度为1.0 nm-2、反应温度为863 K和CH4与O2比为10 mol· L-1时, 甲烷转化率和甲醛选择性分别为1.2%和82.4%。Ohler N和Chempath S等[92, 93]通过EXAFS、Raman光谱和DFT理论计算证明孤立的、含有一个M=O端键的五配位MoOx物种才是甲烷选择性氧化产生甲醛的活性物种, MoOx/SiO2催化剂上甲烷选择性氧化反应机理如图4所示。
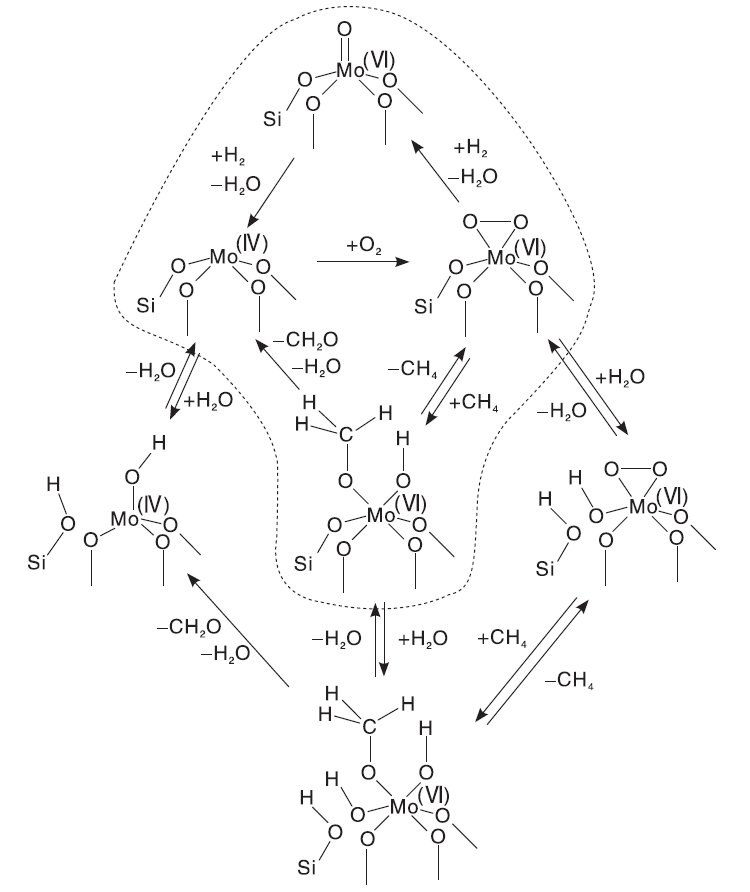 | 图4 SiO2负载孤立MoOx位点上甲烷选择性氧化制甲醛反应机理, 包括有H2O存在所实现的平行反应路径[92]Figure 4 Reaction mechanism of the selective oxidation of CH4 to HCHO over isolated, SiO2-supported MoOx sites, including parallel reaction pathways realized by the presence of H2O[92] |
Mo(Ⅵ )-O物种首先被还原为Mo(Ⅳ )-O物种, 后者在O2气氛中转化变为Mo(Ⅳ )-过氧物种, 然后与CH4反应形成HCHO。值得注意的是, MoOx物种以何种结构参与甲烷选择性氧化反应的机理仍然没有统一定论。Yang W等[94]以孔径更大和热稳定性更高的SBA-15为载体, 研究助剂磷改性对MoOx/SBA-15催化剂性能的影响, 发现磷改性在保持甲醛选择性不变的同时, 可提高甲烷转化率, 但并未说明助剂磷的作用。当反应温度为948 K, Mo质量分数为5%时, 甲醛产率约6%。常规反应条件下, Mo基催化剂的活性仍然较低。近年来, Jurkovi
1.2.2 V基催化剂
甲烷气固相转化通常在较高压力时可获得较好的催化性能, 当反应压力较低(101.325 kPa)时, 催化剂在甲烷选择性氧化反应中起到至关重要的作用[96]。V基催化剂和Mo基催化剂的不同之处在于, Mo基催化剂上甲烷直接被氧化为CO2; 而在V基催化剂上甲烷先被氧化为甲醛, 甲醛在氧化条件下进一步被氧化为CO2。因此, V基催化剂可替代Mo基催化剂应用于甲烷选择性氧化制甲醛反应[23]。在众多的催化剂中, 以SiO2为载体的催化剂性能较好, 也是目前研究较多的一类催化剂。对于负载型V基催化剂, 研究表明高分散的VOx物种才是甲烷选择性氧化生成HCHO的活性物质[97]。在孤立的VOx四面体中, V=O端键被认为是真正的活性位点, 而没有V=O端键的表面VOx物种则导致深度氧化产物的生成; 但也有研究指出, 桥连V-O-Si键也可能参与了反应, 且催化剂表面大量的Si-OH基团也参与了活性位的氧化还原过程, 从CH4分子中提取H原子而使V4+-O中心稳定[97]。尽管活性位的微观配位环境对催化性能的影响研究还不充分, 且存在争议, 但是高分散、孤立VOx四面体的存在仍是V基催化剂具有高活性的关键因素。
Dang T T H等[37]探究了钒源对VOx物种聚合态的影响, 研究发现以VO(acac)2为前驱体主要形成VOx单体, 而以VOSO4为前驱体则形成低聚的VOx物种。高分散的VOx单体有利于甲醇选择性的提高, 当反应温度为873 K时, HCHO空时产率达到5.3 kg· (kg· h)-1, 明显优于以VOSO4为钒源制备的V-MCM-41催化剂[898 K时, HCHO空时产率2.7 kg· (kg· h)-1]。此外, 制备方法和助剂也会影响VOx物种的分散度和催化剂的物化性质, 从而影响催化剂的甲烷选择性氧化性能。Nguyen L D等[97]采用在弱酸性溶液中共缩合的方法制备V-Si催化剂, 该方法制备的催化剂中含有高浓度和高分散的VOx物种, 反应温度为873 K时, HCHO空时产率为6 458 g· (kg· h)-1。Wallis P等[98]制备了V/Ti-SBA-15催化剂, 发现助剂Ti改性有助于VOx物种分散度和还原性的提高, 因此提高了催化剂的甲烷选择性氧化制甲醛性能。Shirmura K等[38]对比研究了助剂Ga加入方式对催化剂性能的影响, 结果表明, 采用顺序浸渍法制备的V(2%)/Ga(0.3%)/SiO2催化剂, 在反应温度863 K和CH4与O2体积比为2条件下, 具有最高的甲醛收率(1.10%), 是未改性V/SiO2催化剂的2.1倍。表征结果表明, 助剂Ga2O3的添加不但没有降低VOx的分散度, 反而提高了VOx的还原性。
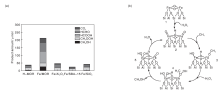
传统的负载型V基催化剂在反应过程中, 分散的VOx物种可能会随着反应的进行而逐渐聚合, 降低催化剂的反应性能。基于此, Yang E等[39]利用先水热、后原子沉积(ALD)的方法制备了一种高效、热稳定性好的SiO2@V2O5@Al2O3催化剂[制备过程如图5(a)所示]。经过50次原子沉积循环后制备的SiO2@V2O5@Al2O3核壳催化剂具有最佳的催化活性, 甲烷转化率为22.2%, 甲醛选择性为57.8%; 反应35 h甲烷转化率和甲醛选择性仍保持稳定[图5(b)]。而没有Al2O3外壳的SiO2@V2O5催化剂在反应过程中会造成结构损失, 且没有催化活性, 表明催化剂的结构对催化性能影响较大。
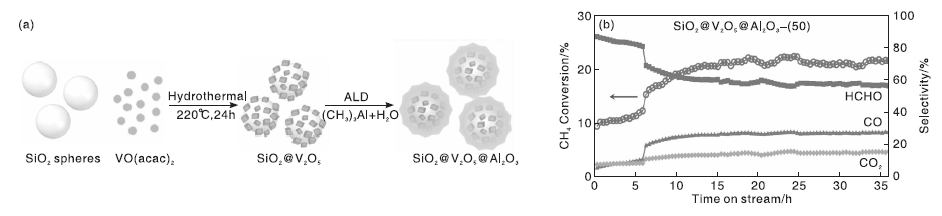 | 图5 SiO2@V2O5@Al2O3核@壳纳米结构的制备过程示意图(a)与甲烷转化率、产物选择性随反应时间变化(b)[39]Figure 5 Schematic preparation process of SiO2@V2O5@Al2O3core@shell nanostructure (a) and CH4 conversion, products selectivity over SiO2@V2O5@Al2O3 catalyst as a function of reaction time on stream[39] |
1.2.3 其他过渡金属催化剂
在过渡金属氧化物表面, 隔离的活性晶格氧原子有助于避免中间产物的深度氧化[99], 因此设计和制备具有孤立活性位的催化剂可以高效地将甲烷氧化为甲醛, 但是活性位的结构和性质受活性金属负载量、制备方法、载体性质和助剂等因素的影响较大。Kobayashi T等[100]很早就发现SiO2载体上高分散、四面体配位的Fe3+物种可以显著提高甲醛产率。以介孔MCM-41为载体, 探究水热法(DHT)和模板离子交换法(TIE)对Fe-MCM-41催化剂性能影响时发现, DHT法使得载体骨架内的四配位Fe-O物种是孤立的, 而TIE法导致在载体中主要形成聚合、八面体配位的氧化铁团簇, 因此TIE法制备的催化剂表现出更高的催化性能[101]。以上研究结果进一步表明, 高分散的Fe位点是获得高HCHO选择性的关键因素。然而, 对于活性Fe物种的结构仍未达成共识。因此, 对FeOx-SiO2进行进一步改性, 以提高其催化性能, 同时研究其结构与催化性能之间的构效关系具有重要的理论意义。助剂P改性的FeOx-SiO2催化剂中, Fe和P之间存在强烈的相互作用, 形成了FePO4纳米颗粒。FePO4纳米团簇中特殊的Fe位点结构(被磷酸基团孤立的Fe3+四面体)有助于甲醛选择性的提高, 当P与Fe物质的量比为0.5、反应温度898 K时, 甲醛单程收率可达到2.4%[102]。
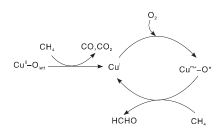
进一步的研究发现, 过渡金属Cu也是潜在的甲烷选择性氧化活性金属。Li Y等[103]研究发现, 高分散的CuOx/SBA-15催化剂以O2为氧化剂能使甲烷选择性氧化为甲醛, 但是Cu负载量对催化性能影响较大。当Cu负载质量分数为0.008%时, HCHO的形成速率为5.6 mol· (mol-Cu· s)-1。脉冲反应和EPR表征结果表明, 与CuⅡ 物种有关的晶格氧与CH4反应生成CO和CO2, CuⅡ 物种还原为CuⅠ ; 而O2在还原的CuⅠ 位点被活化为Cum+-O* 簇, 随之与CH4反应选择生成甲醛, 反应机理如图6所示。同样制备方法也会影响催化剂的甲烷选择性氧化性能, 接枝法制备的CuOx/SBA-15催化剂活性明显高于浸渍法所制备的催化剂。Cu负载量较低时, 催化剂中主要存在孤立的Cu2+物种; 随着Cu负载量增加, 孤立的Cu2+物种减少、低聚的CuOx团簇增加, 甲醛选择性降低[104]。
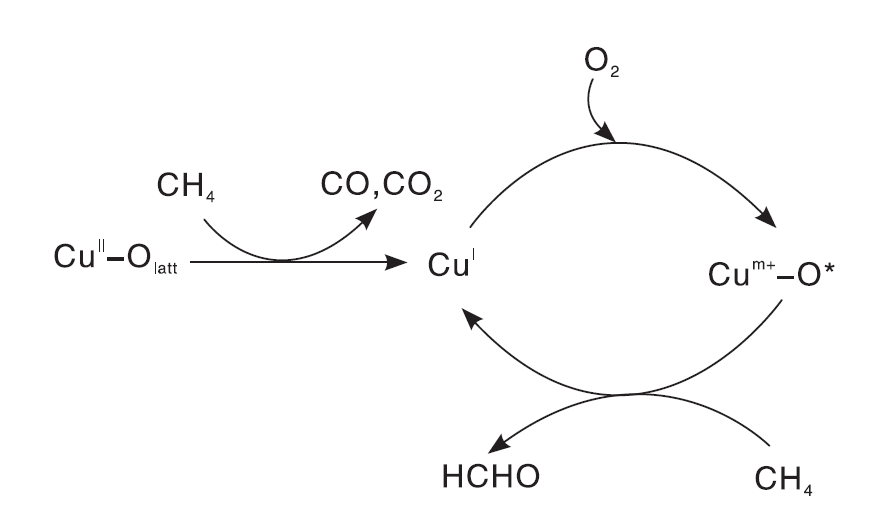 | 图6 CuOx/SBA-16催化剂上CH4选择性氧化为HCHO的反应机理[103]Figure 6 Reaction mechanism for the selective oxidation of CH4 to HCHO over CuOx/SBA-15 catalysts[103] |
甲烷选择性氧化制甲醛气固相催化体系的研究结果表明, 高分散、孤立的活性位点是产生甲醛的关键。但是高分散、孤立的活性位对甲烷的转化程度较低, 且甲烷的活化需要较高的温度和较高的压力, 很难获得较高的甲醛收率; 另外, 对催化活性位结构的研究还不深入, 许多因素(助剂、活性组分含量、载体结构等)对催化活性位结构和性质有显著影响, 因此其可控制备也较难。相信在不久的未来, 这些缺点都可以被克服, 气固相催化体系上甲醛产率定会有所突破。
相比于气固相催化反应, 甲烷液固相催化氧化的研究工作起步晚。液固相催化氧化反应具有设备简单、温度分布均匀、易于控制等优点, 已逐渐成为甲烷选择性氧化反应的重点研究方向。早在20世纪初就有研究指出, Fe2(SO4)3催化剂上甲烷在
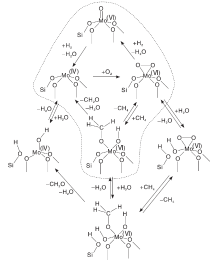
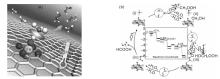
甲烷液固相多相催化氧化体系中, 大部分研究以强酸, 如发烟硫酸、三氟乙酸等为氧化剂, 但是强酸会腐蚀反应器, 从而极大地提高催化反应的运行成本[105, 106]。因此, 开发环境友好的氧化剂和高效的多相催化剂应用于甲烷液固相催化氧化反应具有较好的应用前景。过渡金属具有未充满的价层d轨道, 可以提供空的轨道或孤对电子参与反应, 降低反应的活化能, 从而促进反应进行。Cu、Fe-ZSM-5催化剂在有H2O2存在的水溶液中可以氧化甲烷分子, 但反应需要在323 K、3.05 MPa的间歇式反应器中进行[107]。Fang Z H等[42]采用简单的固相离子交换法制备Fe基催化剂, 发现MOR分子筛是最理想的催化剂载体[图7(a)], 指出位于分子筛骨架外的八面体、二聚Fe3+物种([Fe2(μ -O)2])是初始反应活性位, 并通过DFT计算了催化活性位上甲烷选择性氧化反应的机理, 如图7(b)所示。Cu2+前驱体与Fe3+共存时, Cu2+可有效抑制甲醇的过度氧化, Fe/MOR催化剂上甲醇选择性可达71.3%(反应温度353 K)。Fe/MOR催化剂虽然低温就能使甲烷转变为甲醇, 但是仍然不能实现室温活化甲烷的目的。近来, 包信和课题组[43]报道了石墨烯限域的Fe单原子催化剂在H2O2为氧化剂时, 常温就可以将甲烷直接转化为甲醇和甲醛[图8(a)所示], 且C1含氧化合物选择性高达94%。DFT理论计算和表征结果表明, 催化剂中Fe催化位点的结构为-FeN4-, 反应时液相氧化剂H2O2首先在-FeN4-位点上分解为吸附O物种和H2O, 而此时催化剂表面形成相对稳定的O-FeN4-O物种, 这种物种可以高效活化CH4的C-H键, 从而生成一系列C1含氧化合物[反应过程如图8(b)所示]。
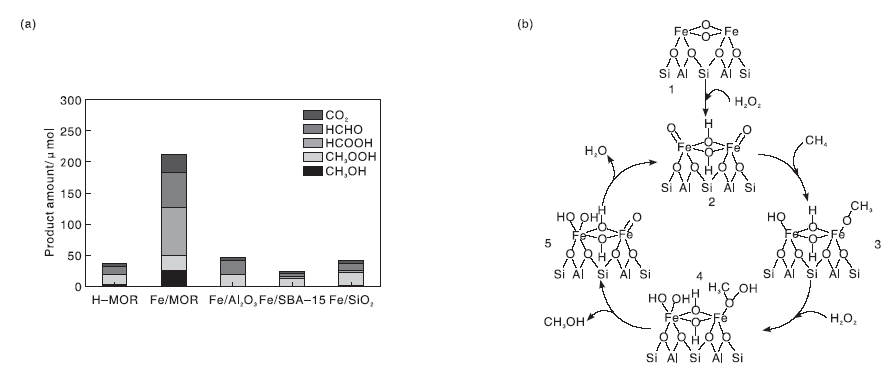 | 图7 不同载体负载质量分数0.5%Fe催化剂上产物选择性(a)和[Fe2(μ -O)2]位点上以H2O2为氧化剂甲烷液相氧化制甲醇的反应机理(b)[42]Figure 7 Production selectivity over Femass fraction of 0.5% loaded on different supports (a) and proposed reaction mechanism for the liquid-phased oxidation of methane to methanol using H2O2 as oxidant over [Fe2(μ -O)2] site[42] |
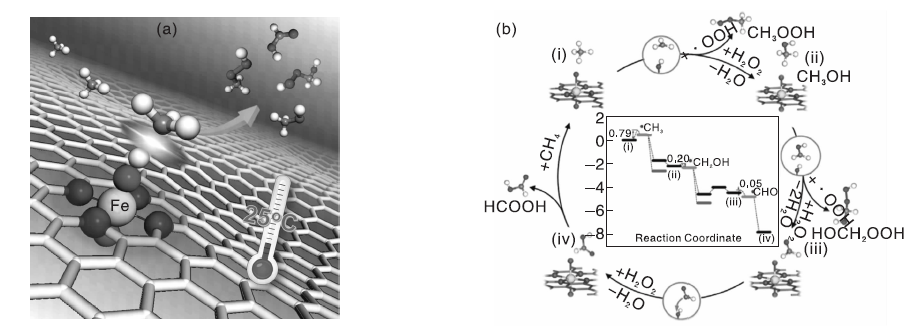 | 图8 FeN4/GN催化剂上甲烷室温转化示意图(a)和反应机理(b)[43]Figure 8 Schematic diagram for room temperature CH4 conversion (a) and reaction mechanism (b) over FeN4/GN catalyst[43] |
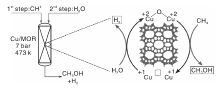
经过长时间的研究, Cu-分子筛已成为最有潜力的甲烷液相催化氧化催化剂。Narsimhan K等[108]以Cu交换MOR分子筛为催化剂、向反应器中同时通入CH4、O2和H2O, 实现了甲烷的持续氧化, 但是甲醇收率较低。该项研究工作具有重要的现实意义— — 第一次证明甲烷可以在连续反应条件下转化为甲醇。从已报道的甲烷液固相催化剂可知, 氧化剂的类型也会影响催化反应的性能。Sushkevich V L等[44]报道了Cu/MOR催化剂能活化甲烷转化为甲醇, 以O2为氧化剂时, 产物为甲醇、甲氧基和过氧化物, 其中甲醇选择性为85%; 以H2O为氧化剂时, 第一轮反应后甲醇选择性仅87%, 随着反应的连续进行, 甲醇选择性和产率提高到97%和0.202 mol· mol-Cu-1。研究表明, H2O不仅可作氧化剂, 还可以使Cu/MOR催化剂的活性位点再生, 反应过程如图9所示。
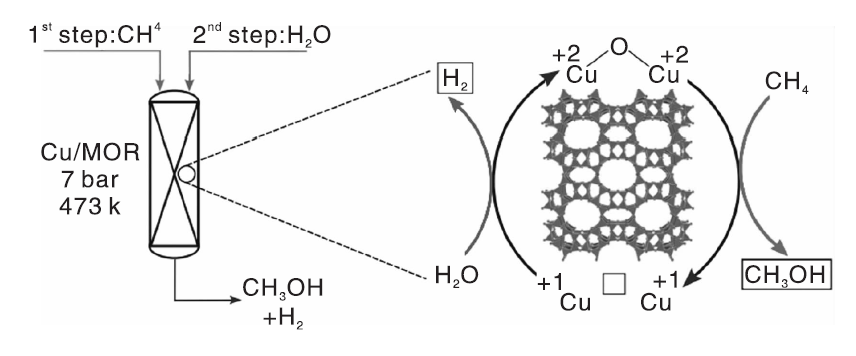 | 图9 H2O为氧化剂, Cu/MOR催化剂上甲烷部分氧化反应示意图[44]Figure 9 Graphical representation of the partial oxidation of CH4 over Cu/MOR catalyst via H2O as oxidant[44] |
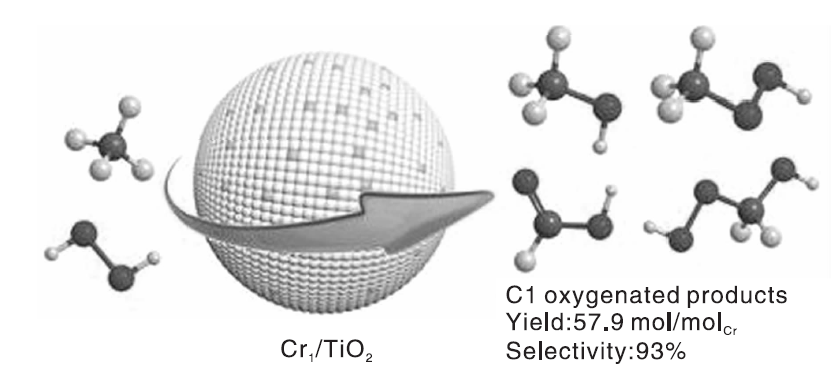
Cu交换MOR催化剂、石墨烯限域的Fe单原子催化剂等应用于甲烷液相催化氧化反应的相继报道说明, 单原子催化剂也能在甲烷选择性氧化反应中大显身手。催化剂制备的两个重要规则:活性位的分离和相间的协同作用。Shen Q K等[45]研究发现, 以H2O2为氧化剂, 不同载体负载的Cr单原子催化剂, 在温和反应条件下展现出优异的甲烷选择性氧化性能。催化剂的高活性归因于单分散的Cr6+物种为催化活性中心, 且Cr原子与TiO2载体之间产生了协同作用(反应过程如图10)。反应温度为323 K时, 连续反应20 h, C1含氧化合物选择性为93%, 产率高达57.9 mol· mol-Cr-1, 明显高于已报道的大多数催化剂。
贵金属催化剂具有高活性、耐高温、抗积炭等优良特性, 成为催化领域的研究热点。从2004年以来, 一直有研究报道Au(Ⅲ )可以活化甲烷分子, 但是并没有获得理想的催化结果[109, 110]。Agarwal N等[46]对负载型Au-Pd胶体纳米粒子进行改进, 制备无载体的Au-Pd纳米粒子催化剂, 以H2O2和O2为共氧化剂, 该催化剂在323 K可将CH4氧化为CH3OH, 其选择性高达92%, 但是该反应过程中
针对甲烷低温氧化反应过程中氧化氢中间体的扩散和O2的扩散效率等问题, 肖丰收课题组[48]提出了“ 分子围栏” 的概念(如图11所示), 用具有疏水表面的沸石包覆AuPd合金纳米颗粒, 可以在低温下高效地将甲烷氧化为甲醇。H2和O2扩散到活性位点原位生成亲水性的H2O2, 而疏水性的硅烷外壳则阻止了H2O2的扩散, 因此活性位点附近H2O2浓度增加, 从而大大提高了甲烷转化效率。当反应温度为343 K时, AuPd@ZSM-5-C16催化剂上甲烷转化率和甲醇选择性分别为17.3%和92%, 甲醇产率高达91.6 mmol· (g-AuPd· h)-1, 是目前最好的研究结果。该结果有助于研究者另辟蹊径去设计和构筑具有高活性的甲烷选择性氧化催化剂[111]。
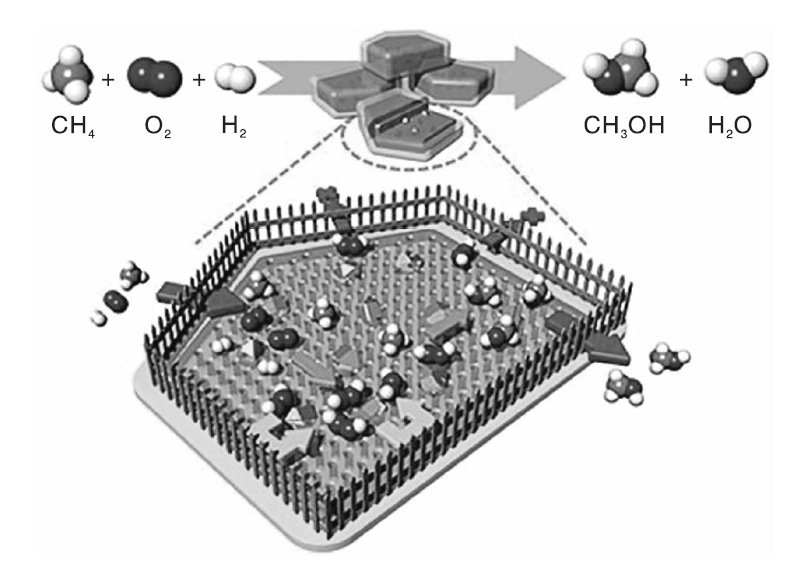 | 图11 “ 分子围栏” 催化剂上甲烷选择性氧化反应示意图[48]Figure 11 Schematic illustration of the selective oxidation of methane over “ molecular fence” catalyst[48] |
近来研究表明, 单原子催化剂(SACs)也可以作为液相甲烷选择性氧化制甲醇的高效催化剂, 其高度隔离的活性位点有利于产物选择性的提高。Bai S X等[49]通过水热法制备CeO2纳米线负载Rh单原子催化剂(SAs Rh-CeO2NWs), 可以在反应温度323 K时(H2O2为氧化剂), 使CH4高效转化为CH3OH, 产率为1 231.7 mmol· (g-Rh· h)-1, 甲醇和甲醛选择为93.9%。与传统浸渍法制备的Rh/CeO2纳米线(Ru主要以团簇形成存在于催化剂中)相比, SAs Rh-CeO2NWs催化剂上CH3OH产率是其6.5倍。反应机理研究发现, · OOH和· OH自由基对产物的形成起着重要作用。SAs Rh-CeO2NWs可以将CH4氧化为· CH3, 其与自由基反应生成产物CH3OH。其研究为甲烷液相催化氧化制甲醇反应提供了新的思路, 也为贵金属SACs催化剂在该领域的应用建立了基础。
贵金属催化剂虽然具有较好的甲烷选择性氧化性能, 但是其价格昂贵, 不利用大规模工业化利用。因此, 寻找廉价的替代催化剂应用于甲烷液相催化氧化反应仍然是一个巨大的挑战。
如何将甲烷分子高效地转化为HCHO或CH3OH等含氧化合物, 并抑制产物深度氧化反应的发生, 仍然是甲烷选择性氧化反应的热点和难点。设计和制备同时具有高活性与高选择性的催化剂是甲烷直接转化研究的关键。多年来, 大量研究者不断努力探索, 已经在甲烷直接转化反应方面取得了重要的研究进展, 但想要实现工业化还有较长的路要走。但目前取得的科研成果对未来开发高效的甲烷选择性氧化催化剂具有重要的借鉴意义。甲烷选择性氧化制含氧化合物反应的研究仍面临着巨大的挑战, 主要包括:
(1) 针对如何保持较高产物选择性的同时, 提高甲烷转化率这一关键问题, 研究者们开展了大量、有效的工作, 但离工业化的要求仍有距离;
(2)有些反应过程的机理尚不明确, 需要更深入探究不同催化剂上甲烷C-H键活化的本质, 建立催化活性位的结构、性质与催化性能之间的构效关系, 从而科学指导高效催化剂的设计与制备;
(3) 气固相催化氧化反应的能耗较高, 且产物收率也较低, 工业化利用受到限制。因此, 需要开发能在温和条件下实现甲烷活化与高效转化的催化剂才能打破这一僵局, 但是难度较高;
(4) 针对甲烷液固相催化氧化反应, 一方面需要实现在环境友好的介质中制备高浓度的甲醇; 另一方面还需要在没有外部能量输入时实现甲烷的活化与转化。只有这样, 才有可能实现甲烷液固相反应催化剂大规模地应用于工业生产。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|